
QuEChERS dSPE EMR-Lipid-UPLC-MS/MS测定蜂蜜中4种硝基咪唑药物残留
2022-05-31 08:13刘虹虹罗达龙
食品安全导刊 2022年13期
王 华,刘虹虹,罗达龙
(梧州市食品药品检验所,广西梧州 543000)
蜂蜜中富含糖类、维生素、氨基酸和多种矿物质等,蜂蜜具有护肝、润肺、增强人体免疫的功能[1]。蜂蜜不仅作为一种营养食品,因其具有一定的药理作用,也被称作医家良药,在临床上用于消化系统、创伤治疗等疾病的治疗[2-3]。因此蜂蜜也成了消费者追捧的产品,在蜂业养殖中为避免蜂群受到疾病感染,会出现抗生素滥用的情况。塞克硝唑、甲硝唑、替硝唑、奥硝唑为《中国药典》2020年版收录的4个具有5-硝基咪唑基本结构的药物,该药物具有抗原虫、抗厌氧菌的作用。在养殖蜜蜂时,养蜂人为避免蜂群出现疾病感染,会使用此类抗生素。由于硝基咪唑类药物具细胞诱变性,有潜在致畸、致癌和致突变的作用,甚至有遗传毒性[5-8]。该类药物在蜂蜜样品中的残留会对食品安全构成直接威胁,且上述4种药物,在药房易于购买获得,因此需加强检测。
蜂蜜在食品工业与药用辅料中有着不可或缺的地位,为保障食品与药用安全,有必要建立一种前处理简单、检测快速、检测限低、定性与定量准确的测试方法。目前,硝基咪唑类药物的相关检测方法为试剂盒快速检测方法、液相色谱法和气相色谱质谱等方法[4]。蜂蜜中存在约70%~80%的糖,糖的去除是实验中的难点。如在前处理中不对糖进行去除,糖会随着流动相带入色谱系统与质谱系统,在色谱系统中会导致进样针与色谱柱的堵塞,也会影响质谱电喷雾过程,使测定的目标物受到基质干扰,产生基质增强或抑制效应,进而导致检测结果不准确。本方法使用了Na2EDTA-磷酸盐缓冲液作为提取溶剂,以QuEChERS dSPE EMR-Lipid为净化剂,SOULAB-ST C18色谱柱为分离手段,运用UPLCMS/MS中的多反应监测模式对蜂蜜样品中的塞克硝唑、甲硝唑、替硝唑和奥硝唑进行检测。
1 材料与方法
1.1 材料与试剂
色谱纯乙腈、甲醇购自于欧姆尼公司;分析纯试剂无水磷酸氢二钠、无水磷酸二氢钠、乙二胺四乙酸二钠盐购自于上海麦克林生化科技有限公司;净化剂QuEChERS dSPE EMR-Lipid购买于安捷伦科技有限公司。实验用水为密理博纯水机制备的去离子水;塞克硝唑、甲硝唑、奥硝唑和替硝唑购自中国食品药品检定研究院,含量均不小于99.0%。
1.2 仪器与设备
超高效液相色谱仪Agilent LC1290,液质联用仪 Agilent-QQQ-6470(Agilent Technologies),超纯水仪(Millipore);涡旋振荡仪(Thermo Fisher Scientific);超声波清洗仪(ELMA);冷冻离心机(SIGMA)。
1.3 实验方法
1.3.1 溶液的配制
(1)提取溶剂的配制。取65 mL浓度为0.2 mol/L的磷酸二氢钠溶液与435 mL浓度为0.2 mol/L的磷酸氢二钠溶液进行混合后,向混合溶液中加入18.6 g乙二胺四乙酸二钠盐试剂,经超声波清洗仪超声溶解后混匀即得Na2EDTA-磷酸盐缓冲溶液。
(2)标准储备液的配制。精密称取标准物质塞克硝唑、甲硝唑、替硝唑、奥硝唑25 mg至4个25 mL容量瓶中,用色谱级甲醇试剂将标准物质溶解,配制得到1 mg/mL的储备液,避光保存于2~8 ℃冰箱。
(3)混合标准液的配制。分别吸取塞克硝唑、甲硝唑、替硝唑及奥硝唑储备液各25 μL到同一个25 mL容量瓶,用色谱纯甲醇试剂进行稀释得到质量浓度为1.0 μg/mL的混合标准液,存放于2~8 ℃冰箱避光保存。
(4)基质标准工作溶液的配制。吸取混合标准液,用阴性样品经前处理后所得到的溶液对混合标准液进行稀释,配制得到的基质标准工作溶液S1~S5浓 度 分 别 为 0.04 ng/mL、0.2 ng/mL、1.0 ng/mL、2.0 ng/mL和5.0 ng/mL,现配现用。
1.3.2 前处理方法
(1)样品制备。若蜂蜜样品中不含有结晶,将其搅拌均匀,备用。如蜂蜜样品中含有结晶,在保证其包装为密封状态下,将样品放置于60 ℃以下的水浴中加热、振荡,等蜂蜜样品全部融化后再把样品搅拌均匀,冷却至室温,即可使用。
(2)样品的提取。称取蜂蜜样品5.00 g到100 mL离心管中,精密加入提取溶剂10 mL,涡旋混合2 min,备用。
(3)样品的净化。吸取提取液5.00 mL至EMRLipid净化管中涡旋混合2 min,精密加入色谱级乙腈试剂5 mL,涡旋混合5 min,在4 ℃下以8 000 r/min离心5 min,吸取上清液0.50 mL到2 mL离心管中,向离心管加入0.50 mL超纯水混合均匀,溶液用0.2 μm有机滤膜过滤,待测定。
1.3.3 测试条件
(1)色谱参数。自动进样器进样量:3 μL;柱温箱恒温温度:35.0 ℃;色谱柱规格:SOULAB-ST C18(100 mm×2.1 mm,1.8 µm);流动相组成:甲醇与0.2%甲酸水溶液;流动相流速:0.35 mL/min;液相色谱流动相梯度:0~0.5 min,10%的甲醇保持0.5 min;0.5~3.5 min,甲醇由10%变化至90%;3.5~4.5 min,90%的甲醇保持1 min;以10%的甲醇比例后运行1.5 min。
(2)质谱参数。正离子模式下喷射流离子源(AJS ESI+),干燥氮气温度(150 ℃);干燥氮气流速(10 L/min);鞘气温度(325 ℃);鞘气流速(10 L/min);正模式喷嘴电压(+500 V);正模式毛细管电压(+4 000 V);雾化器压力(30 Psi)。质谱参数见表1。

表1 化合物质谱参数
2 结果与分析
2.1 实验条件优化
2.1.1 提取及净化方法的选择
蜂蜜中的主要成分是糖分,约占蜂蜜的70%~80%,前处理中需使用净化方法去除糖。如去除糖不完全,会导致色谱柱寿命下降,离子源喷雾针中积累碳化物,同时也会影响待测组分的离子化效率,影响检测方法的灵敏度。硝基咪唑类化合物在弱碱条件下以游离分子的形式存在,故本实验采用偏碱性的Na2EDTA-磷酸缓冲溶液对蜂蜜样品进行提取,使样品得到更好的分散性以提高目标物的提取效率,提取溶剂中含有Na2EDTA、磷酸盐,Na2EDTA在提取液中可与样品中含有的金属离子进行螯合,减少金属离子催化导致待测组分的降解。糖溶解于磷酸盐水溶液中,样品经EMR-Lipid净化后使用乙腈把待测组分萃取到乙腈层,使目标物与糖分别在不同的溶剂层,以达到净化效果。
2.1.2 稀释溶剂优化
液相色谱在分离过程中会存在一定的溶剂效应,如上机溶液选择不合适,会影响物质的保留与色谱峰的峰形,进而影响物质在质谱的响应强度。基于此,本研究用阴性样品经前处理后得到的乙腈层溶液进行稀释,制成含80%乙腈、50%乙腈的水溶液。分别使用乙腈、80%乙腈的水溶液、50%乙腈的水溶液将混合标准溶液配制成待测组分的浓度为5 ng/mL的标准液,上机测试,记录色谱峰的拖尾因子。在使用乙腈、80%乙腈配制的标准液中,甲硝唑与替硝唑色谱峰产生前沿峰,拖尾因子为0.90~0.93;而使用50%乙腈水溶液配制的标准液中,所得物质的色谱峰峰形对称且尖锐,拖尾因子为1.01~1.03,符合拖尾因子0.95~1.05的要求。待测组分溶于乙腈体系中与流动相极性相差较大,产生溶剂效应,进而使待测组分的峰形受到影响。对此为消除溶剂效应,本方法中使用含50%乙腈的空白基质提取液作为标准溶液的稀释液。
2.1.3 色谱条件优化
(1)色谱柱选择。色谱柱在实验中对于样品的分离非常重要,为确保待测组分在色谱柱上有较好的保留与分离效果,比较了柱1 SOULAB-SH C1(8150 mm×4.6 mm,3 µm)、柱 2 SOULAB-AM C18(150 mm×3.0 mm,3 µm)、 柱 3 SOULAB-ST C18(100 mm×2.1 mm,1.8 µm)3种色谱柱对4个待测组分的分离效果。使用柱1与柱2这两款色谱柱所需的流动相流量较大,与质谱电喷雾离子化方式不匹配,物质离子化效率较低,所得的质谱响应强度较低。相对于柱1、柱2,使用柱3能使化合物具有更好的分离效果及峰形,小粒径色谱柱具有高柱效且能节省溶剂,适用于UPLC仪器。综合考虑物质保留时间、色谱峰峰形与检测灵敏度,选择柱3作为本实验分析柱。
(2)流动相选择。本文分别比较了使用有机溶剂甲醇、乙腈作为流动相时对所测定的4种化合物分离效果的影响。结果显示,当使用甲醇作为流动相时,化合物在色谱柱上的保留与分离效果优于乙腈。考虑到待测组分需要与杂质进一步分离,故选择甲醇作为该实验中的有机相。由于4个化合物均采用正模式下的电喷雾离子化,在水中加入一定浓度的酸可以促进化合物的电离。故本研究通过比较在水溶液中加入0.1%~0.5%的甲酸,考察对4个化合物的影响。结果显示,随着流动相中甲酸浓度的增大,4种化合物在质谱测定中得到的峰面积略有增大,但基线噪声也明显提高。综合不同浓度的甲酸作为流动相下所测得物质的信噪比与色谱柱pH的耐受性考虑,选择0.2%甲酸作为水相,化合物的色谱图见图1。

图1 4种化合物色谱图
2.2 检出限、定量限与线性关系考察
取S1~S5基质标准工作溶液,按测试条件进行测定,通过仪器测试得到各化合物定量离子的峰面积。以配制的基质标准工作溶液浓度(x)为横坐标,化合物峰面积(y)为纵坐标,得出回归方程;通过在空白样品中加入标准溶液后对样品进行提取、净化来进行确认本方法所能达到的检出限与定量限,使用工作站对信噪比进行计算,当目标峰峰高与基线噪声的比值≥10,即为定量限;当目标峰峰高与基线噪声的比值≥3,即得出检出限。结果表明,4个化合物检出量在0.2~25.0 μg/kg线性良好,相关系数均大于0.995,检出限均为0.06 μg/kg,定量限均为0.2 μg/kg。化合物回归方程与相关系数见表2。

表2 4种化合物回归方程及相关系数
2.3 回收率实验
取空白基质样品进行加标,通过调节标准溶液的加入体积,使添加量达到 0.2 μg/kg、1.0 μg/kg、10.0 μg/kg,每个加标级别进行6份平行加标实验,并同步进行空白测试,使用基质标准工作曲线对4个待测组分进行定量分析,结果见表3。4个化合物的加标回收率为86.2%~97.0%,相对标准偏差为2.4%~6.5%,均小于10%,表明本方法具有较好的回收率与重现性,可满足蜂蜜中的塞克硝唑、甲硝唑、替硝唑和奥硝唑的测定。
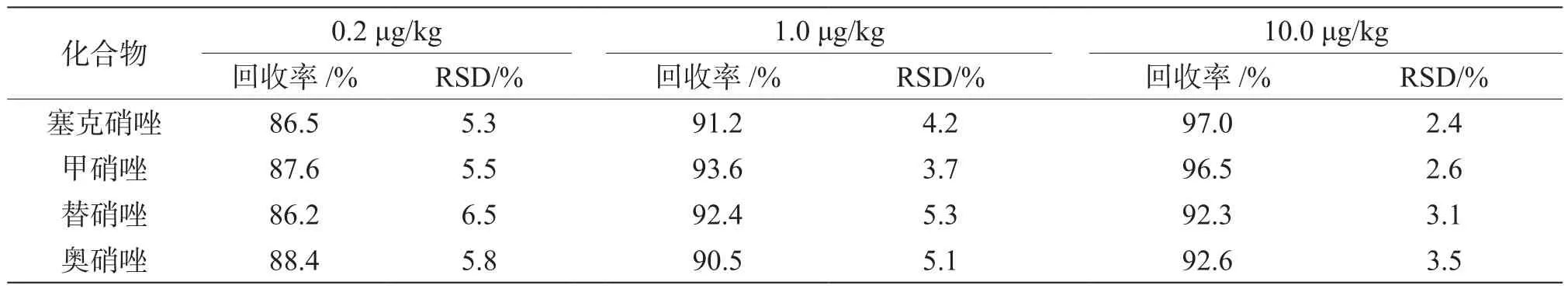
表3 4种基质加标回收率结果(n=6)
2.4 样品的测定
通过购买市场上销售的蜂蜜样品共20批次,按建立的方法进行前处理并测试,结果20批样品中均未检出塞克硝唑、甲硝唑、替硝唑和奥硝唑。
3 结论
本研究建立了QuEChERS dSPE EMR-Lipid-UPLC-MS/MS方法测定蜂蜜中塞克硝唑、甲硝唑、替硝唑和奥硝唑,EMR-Lipid净化剂能有效去除蜂蜜样品中的糖类,用SOULAB-ST C18色谱柱对塞克硝唑、甲硝唑、替硝唑和奥硝唑进行色谱分离,通过质谱联用仪的MRM模式进行检测。方法中使用了空白基质提取液配制标准曲线,相比使用内标物的方法,节省了购买内标物的费用,部分物质的内标物也不容易获得。该方法具有操作简单、快速、定性与定量准确、灵敏度高等特点。本方法使用的净化剂对糖、脂质具有高选择性,能有效去除样品基质中的糖分。糖分的去除能提高待测组分在质谱仪上的离子化效率,减少分析过程中的基质干扰,以提高测试结果的稳定性与可靠性,减少了色谱与质谱的维护成本。
猜你喜欢
中国药学药品知识仓库(2022年7期)2022-05-10
口腔护理用品工业(2021年4期)2021-11-02
原子与分子物理学报(2021年2期)2021-03-29
四川畜牧兽医(2020年7期)2020-07-20
中国医药指南(2017年3期)2017-11-13
老年医学与保健(2017年6期)2017-02-06
老年医学与保健(2017年6期)2017-02-06
中国卫生标准管理(2015年25期)2016-01-14
中国卫生标准管理(2015年24期)2016-01-14
中国卫生标准管理(2015年13期)2015-01-26
