
大豆分离蛋白与大豆磷脂比例对超声乳化亚麻籽油乳液特性的影响
2022-05-29 11:47刘聪慧
中国油脂 2022年5期
刘聪慧,王 欣
(上海理工大学 医疗器械与食品学院,上海 200093)
亚麻籽油富含α-亚麻酸,有助于预防心血管疾病,减少肥胖,降低患癌风险[1]。但亚麻籽油易氧化,在其应用中可能导致食品变质、异味及营养价值降低[2]。O/W乳液是解决食品工业中油脂稳定性问题的缓释输送体系之一,具有安全性高、制备简单等优点[3],因此备受关注。
研究显示,乳化方法对乳液的特性及稳定性有重要影响[4]。常用的乳化方法包括高速剪切、高压均质及超声乳化,其中高速剪切是低能乳化方法,而高压均质和超声乳化是高能乳化方法[5]。近年来超声乳化受到广泛关注,其是基于超声波空化现象产生高温、高压、强剪切力和机械力[6],在较少的表面活性剂下获得具有较好特性、更小液滴的乳液[7]。该方法具有操作简单、成本低、安全、无毒等特点[8]。研究表明,经过不同功率(150~450 W)及时间(12~24 min)的超声处理后,大豆分离蛋白-磷脂稳定的乳液的特性均有所改善[9]。此外,乳化剂组成对乳液特性及稳定性也有重要影响。当前,应用具有优异乳化性能和生物相容性的天然乳化剂已成为趋势。其中,大豆磷脂(SLT)是一种应用广泛且营养价值丰富的天然乳化剂[10]。而大豆分离蛋白(SPI)因其独特的功能特性在乳液的构建中发挥着重要的作用[11]。研究表明,一定比例的SPI和SLT可以在O/W界面共存并产生相互作用,从而有助于乳液特性及稳定性的改善[12]。目前,以SPI和SLT为乳化剂采用超声乳化制备乳液的研究多集中于对超声条件的优化,如:Sui[9]研究了不同超声功率和超声时间对特定比例SPI和SLT稳定的乳液特性和稳定性的影响;江连洲等[13]以大豆蛋白-磷脂酰胆碱复合物为乳化相,葵花籽油为油相,利用超声技术制备乳液,通过响应面优化确定大豆蛋白-磷脂酰胆碱乳液最优超声条件。而对于以SPI-SLT稳定的亚麻籽油乳液特性的研究还相对较少,尤其是超声乳化过程中SPI与SLT比例对亚麻籽油乳液特性的影响更鲜有报道。
本研究以超声制备稳定的亚麻籽油乳液体系为目标,重点就SPI与SLT比例对亚麻籽油乳液的微观结构、水合平均粒径、多分散指数、ζ-电位、分层稳定性、表观黏度及低场核磁共振(LF-NMR)弛豫特性的影响进行研究,以明确其影响规律,确定适宜的SPI与SLT比例,从而为超声制备稳定的亚麻籽油乳液提供一定的参考依据。
1 材料与方法
1.1 实验材料
大豆分离蛋白,分散型,上海国药集团化学试剂有限公司;大豆磷脂,生物技术级,上海麦克林股份有限公司;亚麻籽油,中粮集团有限公司;山梨酸钾,上海麦克林股份有限公司;其他的化学试剂均为分析纯。
JY92-IIDN型超声波细胞破碎仪,宁波新芝生物科技股份有限公司;BX-53型荧光显微镜,日本Olympus公司;Nano Brook 173 Plus型动态光散射仪,美国布鲁克海文仪器公司;Zetasizer Nano-ZS90型粒度分析仪,英国马尔文仪器有限公司;DHR-2型旋转流变仪,美国TA公司;NMI-20型低场核磁共振分析仪,苏州纽迈科技股份有限公司。
1.2 实验方法
1.2.1 SPI和SLT分散液的制备
分别取1.5、1.0、0.5 g SPI加至100 mL去离子水中,另加入50 mg的山梨酸钾抑制微生物的生长,室温搅拌2 h后4℃储存过夜,使其充分水化。调节pH至7.0,4℃下以8 000 r/min 离心15 min,取上清液,即得SPI分散液。向上述SPI分散液中分别加入0.5、1.0、1.5 g的SLT,搅拌2 h使其完全水化,制得SPI与SLT总量为20 mg/mL(以去离子水体积计),SPI与SLT比例分别为3∶1、1∶1、1∶3的分散液。另外,按上述方法分别制备纯SPI分散液和纯SLT分散液。
1.2.2 超声制备亚麻籽油乳液
将2 mL亚麻籽油缓慢加入18 mL的分散液中,置于冰浴中。将超声波细胞破碎仪(最大输出功率900 W,操作频率22 kHz)的钛探头(直径0.686 cm)浸入液面下1 cm处,设定超声功率400 W,超声时间6 min(超声时间4 s,间隔时间6 s),制备O/W乳液,4℃储存。
1.2.3 微观结构观察
采用荧光显微镜观察乳液的微观结构。取10 μL乳液缓慢滴于载玻片上,轻盖盖玻片,防止液滴破裂。在100倍油镜下观察(比例尺10 μm)并采集图像,使用Cell Sens软件观察液滴形态。
1.2.4 粒径的测定
采用动态光散射仪测定乳液的水合平均粒径和多分散指数。分散相的折光指数和吸光度分别设定为1.480和0.001,连续相的折光指数设定为1.330。为避免多重散射现象,测定前用去离子水将乳液样品稀释50倍。在25℃下进行测试。
1.2.5ζ-电位的测定
使用粒度分析仪测定乳液的ζ-电位。为避免多重散射现象,测定前样品用去离子水稀释50倍。取1 mL稀释样品加入电位池(DTS1070)中,在25℃下平衡2 min后进行测定。
1.2.6 分层稳定性的测定
取新鲜乳液样品于5 mL透明玻璃瓶中,旋紧玻璃盖以防样品蒸发。室温避光储存,在储存3、6、12、21 d和30 d观察乳液分层情况。用乳层析指数(IC)表征分层程度,计算公式如下。
(1)
式中:HS为下层水层的高度;HE为乳液总高度。
1.2.7 表观黏度的测定
使用旋转流变仪及其配套的同心圆筒夹具(直径30 mm,间隙4 000 μm)在(25.0±0.1)℃下测定乳液的表观黏度。取15 mL乳液于测试台中,在剪切速率1~100 s-1范围内测定,用TRIOS软件进行数据采集。
1.2.8 LF-NMR分析
采用低场核磁共振分析仪(磁场强度0.53 T,质子共振频率22 MHz)分析乳液的横向弛豫行为。取2.5 mL乳液至核磁试管中(直径为15 mm),32℃预热10 min后检测。选择CPMG序列进行横向弛豫时间(T2)检测。具体参数:重复扫描次数(NS)4次,回波个数(Echo Count)6 000,重复采样等待时间(TW)2 000 ms,采样频率(SW)250 kHz。利用配套的T-Invfit反演拟合软件对弛豫衰减曲线进行拟合,得到样品的T2信息,如各峰的起始弛豫时间(T2i,i=1,2,…,n),绝对峰面积(A2i,i=1,2,…,n),峰面积比例(R2i,i=1,2,…,n),单组分弛豫时间(T2W)等。
1.2.9 统计学分析
所有实验重复3次,结果表示为“平均值±标准差”。利用SPSS 21.0统计分析程序对数据进行ANOVA方差分析,采用Duncan多重极差检验评价差异的显著性(P<0.05为显著性差异)。采用Origin 8.0软件进行数据、图表及图谱分析处理。
2 结果与讨论
2.1 乳液的微观结构及粒径
乳液的液滴大小易影响乳液的物化特性,与乳液的失稳现象如相分离、液滴聚集和油上浮等密切关联[14]。SPI与SLT比例对乳液微观结构的影响见图1,SPI与SLT比例对水合平均粒径及多分散指数的影响见表1。

注:a.仅添加SPI;b.SPI与SLT比例3∶1;c.SPI与SLT比例1∶1;d.SPI与SLT比例1∶3;e.仅添加SLT。
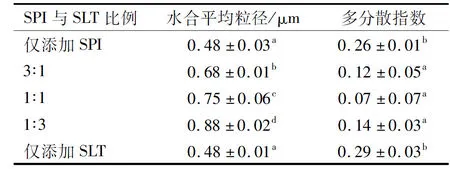
表1 SPI与SLT比例对乳液水合平均粒径及多分散指数的影响
由图1可知,与仅添加SPI或SLT相比,SPI与SLT比例在3∶1~1∶3时所形成的乳液液滴尺寸较大。由表1可知,仅添加SPI或SLT时,乳液的水合平均粒径较小,为0.48 μm,但其粒径分布图(图2)均呈双峰分布,且其多分散指数(PDI)较高,分别为0.26和0.29。当SPI与SLT比例为3∶1~1∶3时,乳液的水合平均粒径在0.68~0.88 μm之间,且随SPI与SLT比例的减小,乳液的水合平均粒径增大;乳液的粒径分布图(见图2)均呈单峰分布,且PDI较低,为0.07~0.14。这是因为当SPI和SLT共同作为乳化剂时,两者可以在O/W界面共存并产生疏水相互作用与静电相互作用,使乳液中液滴分布更均匀[11]。当SPI与SLT比例为1∶1时,乳液的PDI最低,达到0.07,表明其具有更好的均匀性。

图2 SPI与SLT比例对乳液粒径分布的影响
2.2 乳液的ζ-电位
ζ-电位是表征乳液稳定性的一个重要指标,其绝对值越高,粒子间的排斥力越大,反之粒子间越倾向于相互吸引而发生聚集[15]。SPI与SLT比例对乳液ζ-电位的影响如图3所示。
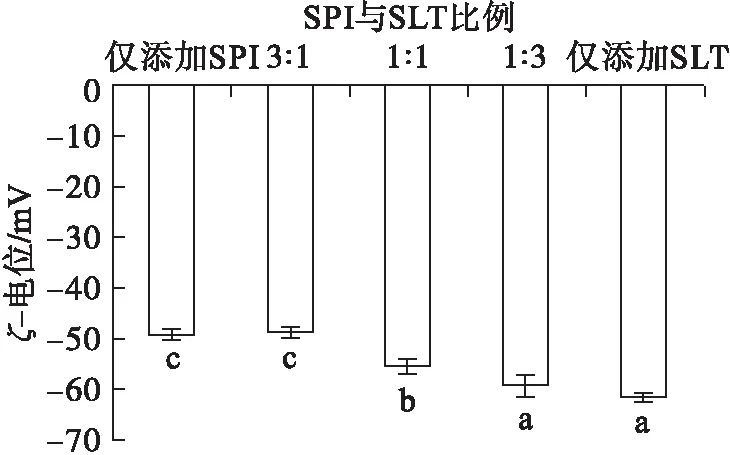
注:不同的小写字母表示有显著性差异(P<0.05)。
由图3可知,所有样品的ζ-电位均为负值,表明液滴周围存在阴离子[6]。随着SPI与SLT比例的减小,ζ-电位绝对值增加,且总体大于仅添加SPI的。这可能是由于SPI与SLT之间的相互作用改变了蛋白质表面的电荷分布。研究表明,SPI与SLT之间的疏水相互作用可以打开蛋白质构象,暴露出更多带负电的残基,粒子间的静电斥力增强,使ζ-电位的绝对值升高[16]。增加的静电斥力可以破坏蛋白质聚集体,提高乳液的稳定性。而当仅添加SLT时乳液电位绝对值较不同SPI与SLT比例乳液的高,这可能与磷脂中含有带负电荷的磷酸基团相关[17]。
2.3 乳液的分层稳定性
乳层析指数表示油滴在乳液中从水相聚集和分散的程度,较低的乳层析指数意味着乳液更为稳定[9]。不同储存时间下SPI与SLT比例对乳液乳层析指数的影响如图4所示。

注:不同小写字母表示相同储存时间下不同SPI与SLT比例乳液间有显著性差异(P<0.05)。
由图4可知,在30 d的储存过程中,各乳液体系乳层析指数均增加。储存3~12 d时,随SPI与SLT比例减小,乳液的乳层析指数增加,储存3 d时,当SPI添加量从仅添加SPI下降至SPI与SLT比例1∶3时乳层析指数从2.85%显著增加至6.10%,表明SPI与SLT比例为1∶3时,乳液的分层稳定性最差,且差于仅添加SPI或SLT的。储存21 d时,随着SPI与SLT比例的减小,乳液的乳层析指数变化不大,且仅添加SLT稳定的乳液的乳层析指数显著大于其他组,乳液稳定性最差。整体而言,仅添加SPI时乳液的乳层析指数最低。已有研究表明[18],表面活性剂稳定的乳液容易絮凝和聚集,而Pickering乳液具有独特的优势抵抗絮凝和聚集。另一方面,尽管磷脂与大豆蛋白之间的疏水和静电相互作用形成的界面吸附膜有助于提高乳液的稳定性[19],但二者之间的比例对体系稳定性有重要影响。当体系中SLT过量时,有可能产生竞争吸附效应,过量的SLT取代O/W界面上的SPI,导致其聚集,从而使乳液稳定性下降。
2.4 表观黏度
作为乳液稳定性的重要表征方法之一,表观黏度主要取决于液滴之间的相互作用,且能反映蛋白质之间及蛋白质与水的相互作用[20]。SPI与SLT比例对乳液表观黏度的影响如图5所示。

图5 SPI与SLT比例对乳液表观黏度的影响
由图5可知,在剪切速率为1~100 s-1的范围内,5种乳液的表观黏度均维持在10 mPa·s以下,表明其黏度较低、质地偏稀、流动性大。在1~5 s-1的低剪切速率时,5种乳液表观黏度随剪切速率的增加而降低,表现出剪切变稀的非牛顿流体特征,这可能是由于在低剪切速率下,乳液发生变形并被瓦解;在剪切速率大于5 s-1时,5种乳液表观黏度保持稳定,表现为牛顿流体特征,这可能是由于剪切速率增加时,乳液瓦解速率与絮体再形成速率相当,这与王中江等[21]报道的趋势类似。另外,随SPI与SLT比例下降,乳液的表观黏度上升,这可能与体系中黏度较大的SLT量增加,从而增加了体系的黏度有关。
2.5 乳液的LF-NMR弛豫行为
LF-NMR弛豫特性可反映乳液体系中氢质子的运动状态[22]。其中,T2对氢质子运动敏感,是反映乳液形成过程中水和油分子运动的一个重要特征[23]。氢质子受到的束缚越大,T2越短[24]。乳液衰减曲线可反映样品的衰减速度,单组分弛豫时间(T2W)用于反映样品的总体弛豫分布,相当于最大信号衰减到63%时所需的时间[25]。SPI与SLT比例对乳液衰减曲线的影响如图6所示。
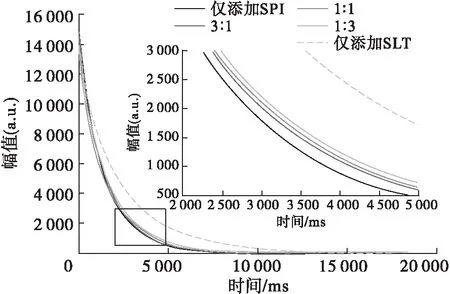
图6 SPI与SLT比例对乳液衰减曲线的影响
由图6可知,当SPI与SLT比例在3∶1~1∶3时,5种乳液的衰减过程有明显区别。当仅添加SPI时,乳液的衰减曲线具有最大的衰减速率且最先到达衰减终点。总体来说,当SPI与SLT比例在3∶1~1∶3时,随SPI与SLT比例减小,乳液的衰减过程逐渐变慢。而仅添加SLT时,乳液的衰减过程明显减缓,到达衰减终点所需时间最长。这也反映在T2W上,见表2。

表2 不同SPI与SLT比例的乳液的T2W
由表2可以看出,5种乳液的T2W介于1 070~1 783 ms之间,且随着SPI与SLT比例的减小,乳液的T2W逐渐增加,表明体系中氢质子所受束缚力逐渐减小。SPI可以在O/W界面形成膜,从而限制氢质子的自由度[26],随SPI与SLT比例减小,O/W界面上覆盖的蛋白颗粒减少,其对水和油中氢质子的束缚力降低,因此T2W相对增大。
SPI与SLT比例对亚麻籽油乳液多组分弛豫图谱的影响如图7所示,图中每个弛豫峰代表一种状态的氢质子,可用于反映特定氢质子的弛豫信息。
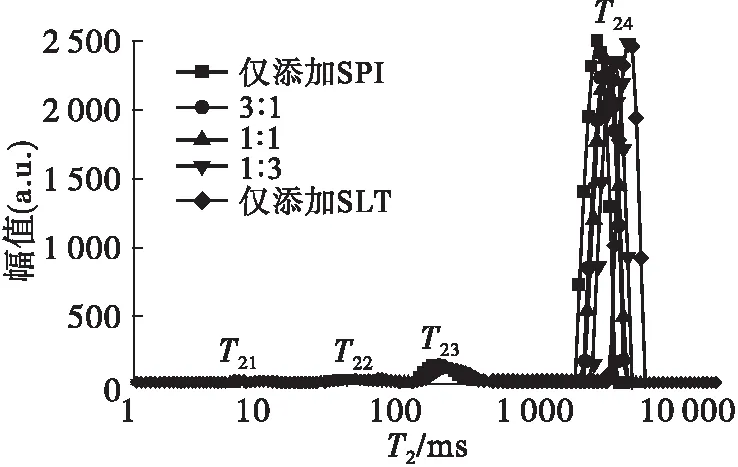
图7 SPI与SLT比例对乳液多组分弛豫图谱的影响
由图7可知,当仅添加SPI时,乳液的多组分弛豫图谱分别在19.34 ms(T22峰的起始时间)、77.53 ms(T23峰的起始时间)和1 084.37 ms(T24峰的起始时间)左右出现了3个弛豫峰,说明体系中存在3种不同类型的氢质子。然而,仅添加SLT时,乳液的多组分弛豫图谱在5.94 ms左右(T21峰的起始时间)又出现了一个新的弛豫峰。当SPI与SLT比例在3∶1~1∶3之间时,T21峰也出现在相应的多组分弛豫图谱上,但峰面积比较小。已有研究表明,该弛豫峰与磷脂双分子层中的氢质子有关[27]。在其他3个弛豫峰中,T24占比最大,该峰与水中的氢质子响应有关[28]。而T22和T23代表油中的氢质子[19]。随SPI与SLT比例减小,T23和T24右移,这可能是由于对SPI稳定的Pickering乳液而言,通过SPI固体颗粒覆盖在界面上形成膜来稳定O/W界面时,分子间的相互作用较强,氢质子的移动和旋转空间相对较小,氢质子自由度较低,因此其T2较短。而SLT作为一种小分子表面活性剂,主要通过降低O/W界面张力形成乳液[29],SLT为氢质子提供了更大的旋转和运动空间,因此其T2较长。因此,乳液体系中的SLT越多,氢质子所受的束缚力越弱,其T2越长。
3 结 论
对于超声乳化制备的亚麻籽油乳液,SPI与SLT比例对亚麻籽油的乳液特性有明显影响。随SPI与SLT比例减小,乳液的水合平均粒径增大,多分散指数先减小后增大,ζ-电位绝对值、表观黏度以及乳层析指数总体增加,T2弛豫图谱右移。当SPI与SLT比例为1∶1时,乳液体系的多分散指数最低(0.07±0.07)且粒径呈单峰分布,ζ-电位绝对值较高,乳层析指数较低,体系中氢质子所受的束缚力较大,表观黏度较大,得到的亚麻籽油乳液体系相对更均匀、稳定。研究结果可为乳液体系的构建提供一定的参考。
猜你喜欢
食品工业科技(2022年21期)2022-10-27
中国粮油学报(2022年8期)2022-09-28
物探与化探(2022年4期)2022-08-26
土壤学报(2022年1期)2022-03-08
发明与创新(2022年1期)2022-02-20
林业科技(2020年3期)2021-01-21
种子(2020年4期)2020-05-20
商品与质量(2018年38期)2018-04-16
散文诗世界(2017年3期)2017-11-13
家庭百事通·健康一点通(2016年7期)2016-08-04
