
特异性睡眠脑电频率在阿尔茨海默病中的研究进展
2022-05-27 10:39刘善雯刘春风胡华
实用老年医学 2022年5期
刘善雯 刘春风 胡华
随着人口老龄化,AD发病率逐年增加,每增加5岁,AD发病率就增长近一倍[1]。大量研究表明,睡眠障碍是AD的常见症状,可以贯穿各阶段,尤其在病程后期睡眠障碍更严重。值得注意的是,失眠、阻塞性睡眠呼吸暂停综合征(obstructive sleep apnea syndrome, OSAS)、睡眠片段化、白天嗜睡等睡眠障碍在AD临床前期[2],即轻度认知功能障碍(mild cognitive impairment,MCI)、主观认知功能减退(subjective cognitive decline,SCD)甚至健康时可能就已存在。有睡眠障碍的病人发展成AD的风险会提高51%,尤其是遗忘性轻度认知功能障碍(amnestic mild cognitive impairment,aMCI)病人,比健康人有10倍以上概率可能发展成AD[3]。上述这些流行病学资料提示我们睡眠障碍可能参与AD发生发展的始动环节。
1 睡眠障碍可能是AD的起因之一
AD睡眠障碍表现形式多样复杂,以失眠、睡眠觉醒节律紊乱、睡眠片段化、睡眠呼吸障碍、白天嗜睡等较为常见。传统观点认为睡眠障碍继发于AD,其主要原因是随着AD进展,不断沉积的β淀粉样蛋白(amyloidβ-protein,Aβ)和形成的磷酸化tau蛋白(phosphory protein tau, p-tau)通过炎症反应等机制损伤睡眠调节相关脑区[4],如基底前脑、下丘脑、丘脑等,引起这些脑区神经突触功能障碍,进而出现上述不同类型的睡眠紊乱症状。
但近年来相关研究提示,睡眠障碍不仅继发于AD,可能是AD的起因。一项基于社区的前瞻性研究,经过长达6年的随访,发现睡眠片段化发生率高的人群患AD的风险较正常对照组高出1.5倍[5]。另一项随访时间更长(长达40年),样本更大(1574例)的观察研究表明:有睡眠障碍的人群患AD的风险要高出1.51倍。诸多类似研究均提示:睡眠障碍往往会在AD病人认知功能减退之前就已出现,睡眠障碍可能是AD的起因之一[6]。
2 睡眠障碍参与AD的可能机制
2.1 神经病理学机制 AD神经病理学金标准是神经细胞外Aβ沉积形成的老年斑、p-tau所致的细胞内神经纤维缠结(neurofibrillary tangles,NFTs)。Aβ是指不溶性的Aβ40和Aβ42。p-tau与微管蛋白形成的异常双螺旋形态,会聚集形成NFTs,损害神经元微管结构,导致死亡。
研究发现,在认知功能正常的人群中,有Aβ沉积者比无Aβ沉积者睡眠质量更差,入睡后醒来次数增加[7];Aβ沉积与更短的睡眠时长、更多的片段化睡眠、更低的睡眠质量相关,睡眠障碍甚至能预测Aβ沉积和p-tau形成[8]。
生理状态下脑内Aβ水平呈现昼夜波动性:清醒时分泌增加,睡眠时分泌减少。睡眠能有效清除脑脊液(cerebrospinal fluid,CSF)中Aβ42和p-tau,随着年龄增加,清除功能会生理性地减少,当睡眠剥夺时,这种清除功能会明显下降[9]。低质量睡眠造成觉醒时间增长,皮质神经元活动增加, Aβ大量释放与沉积会破坏与睡眠周期变化有关的神经环路,导致睡眠结构紊乱;睡眠结构的紊乱反过来又会造成Aβ的沉积,进一步影响睡眠。可见,睡眠障碍会增加Aβ沉积、清除减少,二者之间有相互促进关系。
2.2 神经递质调节紊乱 睡眠-觉醒周期是由基底前脑、丘脑、下丘脑、脑干等调节,通过上行网状抑制或激活系统释放调节觉醒的兴奋性或抑制性神经递质,对大脑皮层产生易化或抑制,对学习记忆的形成与巩固有重要作用。乙酰胆碱(acetylcholine,Ach)可以维持睡眠觉醒和大脑皮质活动、保持身体警觉。研究证明,快速眼动(rapid eye movements,REM)期睡眠与Ach调节觉醒的高效性密切相关,REM期乙酰胆碱能神经元可以促进神经可塑性,参与记忆形成[10]。5-羟色胺(5-hydroxytryptamine,5-HT)是上行激动系统的组成部分之一,REM期睡眠剥夺会导致额叶5-HT水平增高,造成大鼠学习记忆能力下降,推断REM期睡眠对维持5-羟色胺能神经元正常活动至关重要[11]。
2.3 神经影像学 从影像学角度来看,睡眠障碍的人群与AD病人许多脑区存在相似变化。一项对45~75岁认知功能正常人群进行的研究发现,失眠病人在AD相关脑区(如左角回、双侧额上回、丘脑和右海马体)灰质体积更小,左尾状核体积更大,右半球白质束的平均和轴向扩散率降低,而这种改变在携带ApoE-ε4的失眠人群中往往更常见[12]。认知功能未受损的失眠病人,海马、楔前叶、杏仁核和扣带回的灰质体积减少,皮质萎缩程度增加。故推断认为,睡眠障碍在AD诊断之前,可能就在相关脑区发生了类似的大脑结构性改变。此外,主诉有睡眠障碍的MCI病人比正常人脑白质高信号体积更显著,表明睡眠障碍可能通过损害大脑血管、破坏血脑屏障、影响神经元活动,导致认知功能不同程度的受损[13]。
3 从睡眠脑电结构角度探讨AD发生机制
1949年加拿大生理心理学家赫布提出,记忆的痕迹最先以神经冲动的振荡形式存在,这就是最初的短时记忆,这种神经冲动的振荡改变突触的连接形式、大脑化学或结构改变等,成为长时记忆。早期记忆的形成主要为海马依赖型;之后记忆存储和巩固主要依赖于新皮层不同脑区间建立的相互联系。
正常的睡眠结构对记忆的形成、维持、巩固有着至关重要的作用。清醒状态下,外部信息的输入由分布在大脑新皮层和海马结构中的神经元组合编码形成新记忆。睡眠期间,没有了外部信息输入,海马-新皮层、丘脑-皮层环路重复激活、加强神经连接并重组记忆,将清醒时形成的一部分新记忆整合到原先记忆中,建立更强的海马-皮质间连接,形成长时记忆,同时也清除了另一部分弱的记忆。见图1[14]。
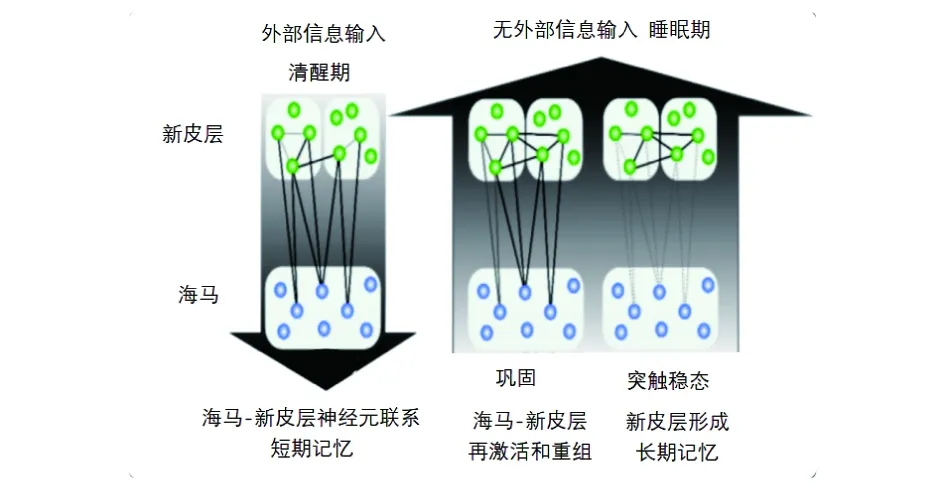
图1 主动系统巩固假说
神经元间的信息转导离不开睡眠期有序的脑电波耦合。睡眠脑电结构包括REM和非快速眼动 (non-rapid eye movements, NREM)睡眠间的循环交替。NREM睡眠分为浅睡眠[NREM 1期(N1期)、NREM 2期(N2期)]和深睡眠[NREM 3期(N3期)、NREM 4期(N4期)]。N2期有一个典型的丘脑-皮质再激活,表现为12~15 Hz突然爆发的振荡活动,称为睡眠纺锤波,睡眠纺锤波也发生在N3期,但经常被慢波振荡(slow oscillations,SO)所掩盖[15]。N3期的特征是1~4 Hz的慢波和<1 Hz的SO,代表了慢波睡眠(slow wave sleep, SWS),在这一阶段,海马体和新皮层之间的相互作用,参与记忆维持、巩固。N3期也出现海马尖波,被称为高频振荡(120~200 Hz)的高度同步尖峰间歇模式,代表了海马记忆表征的重新激活,并被认为是情景记忆的认知生物标志物[16]。REM睡眠的特征是出现在海马体的θ活动(4~8 Hz),加上γ活动(30~120 Hz),它可以调节NREM睡眠中记忆通路的整合和重组[17]。其中,NREM睡眠的振荡波合乎时宜的耦合对海马、新皮层和其他结构间的正确信息转导至关重要。SO-睡眠纺锤体耦合可促进突触的可塑性,维持及巩固记忆。同样,海马尖波准确耦合到睡眠纺锤体,可以加强情景记忆形成。脑电波正确耦合将重新激活的海马信息通过丘脑传输到新皮层,并促进突触整合,形成长时记忆。见图2[14]。
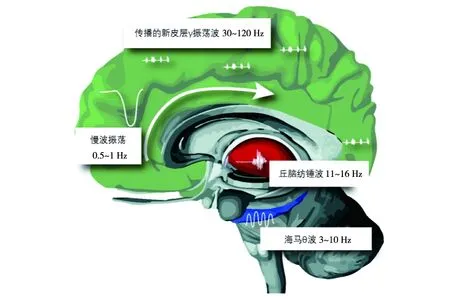
图2 睡眠脑电频率耦合模型
现有研究证实,AD睡眠障碍病人在睡眠脑电结构上存在着特异性改变。比如睡眠障碍类型中最常见的失眠,发现失眠病人下丘脑、丘脑、上行网状激活系统、岛叶皮质、杏仁核、海马、前扣带皮质和内侧前额叶皮质脑电活动下降,在情绪和认知表现上更差。失眠病人SWS和REM睡眠减少、持续时间缩短,短暂觉醒和微觉醒频率增加,进入REM期的潜伏期增加,提示其觉醒过度活跃[18]。这些变化,特别是SWS的减少,导致了睡眠依赖的陈述性记忆巩固的损害。
4 预测AD的几种脑电频率及机制
4.1 脑电图(electroencephalogram,EEG) 早期研究表明,正常衰老人群深睡眠减少、觉醒频率及时间增加,AD病人更为显著,伴有睡眠纺锤波形成不良或缺失[19]。一项通过建立3、4、6个月龄的AD小鼠来研究EEG变化的研究中发现,在12 h黑暗期,4月龄、6月龄AD小鼠比3月龄AD小鼠觉醒时间更少,NREM睡眠时间更长,提示随着疾病发展会逐渐有嗜睡倾向[6]。6月龄或更大月龄AD小鼠在AD病理标志物出现之前,认知功能即已出现减退,说明睡眠结构的改变早于AD病理形成和认知功能的下降。
4.2 NREM <1 Hz SO和1~2 Hz的SWS减少 Mander等[20]的研究表明,在MCI、AD、健康老年人中,SO减少会导致更多的Aβ沉积。最早沉积在内侧前额叶皮质的Aβ水平升高与<1 Hz SO显著相关,其他产生SO的脑区(如外侧前额叶皮层、后扣带、楔前叶),CSF可溶性Aβ42水平升高,且随着AD病情加重,逐渐累及海马,所以NREM <1 Hz SO的减少可能是一种临床前AD和Aβ沉积的预测因子。
同样,在无症状或轻度AD病人中发现,1~2 Hz的SWS减少与CSF p-tau/Aβ42升高、灰质神经元丢失显著相关[21]。PET探测到tau蛋白多形成在产生1~2 Hz的SWS的脑区(如嗅皮层、杏仁核、内嗅、海马旁、顶下叶),所以1~2 Hz的SWS减少也可能是临床前AD和tau形成的预测因子。
4.3 N2期睡眠纺锤体减少 一项针对健康老年人的研究发现,睡眠纺锤体数量减少与CSF t-tau增加显著相关,与CSF Aβ42、p-tau升高呈轻度相关,睡眠质量或持续时间、额叶SWS的减少与CSF t-tau无相关性[22],证实睡眠纺锤体预测CSF t-tau的特异性。鉴于睡眠纺锤体在记忆巩固和神经可塑性方面的作用,还与更好的认知表现相关,纺锤波可能也是一种临床前AD和CSF tau形成的预测因子。
4.4 N3期 SO-睡眠纺锤体耦合减少 研究发现,在调整年龄、性别、OSAS等因素后,SO-睡眠纺锤体耦合仅与颞中叶形成的tau蛋白相关,与皮层Aβ无相关性。可能是因为SO-睡眠纺锤体的耦合参与依赖睡眠的海马记忆过程,其破坏会加速颞中叶tau蛋白的形成[23]。
上述主要是针对NREM期睡眠特异性脑电频率与AD之间的关联,尚未检索到AD病人REM期变化的相关研究,鉴于现有临床研究中观察到REM期睡眠减少的MCI和AD病人的认知测试结果更差[24],或许可以进一步探索REM期的特征性脑电频率与AD二者之间的关系。综上所述,笔者将现有可能预测AD的脑电频率及对应病理标志物定位,汇总如下。见表1。
5 针对睡眠脑电频率进行AD早期干预
调节睡眠是一种有效的干预途径,尤其在最初因睡眠紊乱导致依赖海马的记忆损害及Aβ沉积的病人中,早期纠正睡眠紊乱可能是有效且重要的措施。目前,有几种候选方法可以实现增强NREM期睡眠效率,特别是针对<1 Hz NREM SO。
5.1 <1 Hz NREM SO内的经颅直流电刺激(transcranial direct current stimulation,tDCS) tDCS可以使年轻人的夜间睡眠依赖性记忆提高2倍[25]。在老年人及颞叶癫痫、注意力缺陷多动障碍病人,甚至在啮齿动物中均证明了<1 Hz NREM SO内的 tDCS的记忆巩固作用[26]。然而也有报道在年轻人和老年人中,<1 Hz NREM SO tDCS治疗并未发现有增强记忆的作用[27],因此,对该治疗方法还需要进一步研究。
5.2 SWS期听觉闭环刺激 研究发现睡眠过程中给予缓慢、有节奏的动觉刺激(如婴儿摇床),可以显著增加年轻人的低频NREM SO[28],但该研究并未进行记忆评估,这种睡眠改善是否会增强记忆尚未明确,但该研究也不失为一种可期待的研究方向。
5.3 认知行为疗法(cognitive behavioral therapy for chronic insomnia,CBT-i) CBT-i是一种非药理、非侵入性的方法,可有效改善慢性失眠病人的睡眠质量[29]。由于失眠在衰老、MCI和AD病人中更为突出,并可增加患AD的风险,因此CBT-i是另一种可采取的有效干预措施。目前尚不清楚CBT-i是否能改善睡眠脑电结构,包括<1 Hz NREM SO,所以同样值得被关注。
6 结语与展望
综上所述,我们需关注AD睡眠脑电结构特异性变化,可以通过建立睡眠脑电结构与影像学、CSF等病理学联合诊断模型,并结合诸如tDCS等睡眠脑电结构干预措施,通过调节特异睡眠脑电频率来进一步对AD病理机制深入研究,或许可以为AD早识别、早诊断、早治疗带来新的方向。此外,现阶段对AD与其他类型痴呆在睡眠脑电结构的区别研究尚少,因此我们未来可以探讨AD和其他痴呆类型特异性睡眠脑电频率的差异,以辅助早期识别不同痴呆类型。
猜你喜欢
中华实验眼科杂志(2022年6期)2022-11-15
心理学报(2022年10期)2022-10-12
西北大学学报(自然科学版)(2022年4期)2022-07-20
心理学报(2022年3期)2022-03-08
心理学报(2022年1期)2022-01-21
心理学报(2021年8期)2021-08-11
首都体育学院学报(2019年5期)2019-10-18
中学科技(2018年9期)2018-12-19
中国当代医药(2017年17期)2017-07-25
健康管理(2017年3期)2017-04-20
