
氮掺杂石墨烯原位生长碳纳米管复合过渡金属催化剂的制备及电催化性能
2022-05-23 04:54李生娟于沺沺李田成薛裕华
材料工程 2022年4期
张 婷,李生娟,吉 莹,于沺沺,李田成,薛裕华
(上海理工大学 材料科学与工程学院,上海 200093)
近年来,可充电锌空气电池因安全性高、能量密度大、资源丰富、环境友好而被认为是一种很有前途的储能装置[1]。然而,在反应过程中,动力学缓慢、过电位大等缺点降低了锌空气电池空气阴极上氧还原/氧析出(ORR/OER)的反应效率,从而限制其规模化应用。目前主要应用于ORR的贵金属和OER的贵金属氧化物催化剂分别有Pt,Pd和RuO2,IrO2等,但这些催化剂稳定性差、成本高、来源稀少、催化活性单一。因此,目前科研人员致力于开发高效、低成本、高性能的双功能催化剂。一维碳纳米管(CNTs)具有比表面积大、导电性好等优点,二维石墨烯(GO)具有大的比表面积、优异的导电性和良好的电化学稳定性等[2-3],将两者结合可以得到具有优异导电性及电化学稳定性的三维复合结构。然而纯碳材料催化活性较低,在进行催化反应时不可避免发生碳腐蚀[4],从而加剧催化剂的聚集。化学掺杂杂原子(如氮、磷、硼、硫等)是一种从本质上改变基体材料性质的有效方法,可以进一步提高其催化活性及稳定性。在元素周期表中,氮与碳相邻,具有类似的原子半径和电负性,因此,氮更容易融入碳晶格中,产生额外的孤对电子,加速电子的转移,改善碳载体的电子性质,从而提高ORR活性。Liu等[5]报道了N掺杂碳纳米管(NCNTs)中氮原子的掺杂可以有效地诱导碳材料中原子电荷密度和自旋密度的重新分布,并调节碳原子的O2吸附/解吸能,从而提高ORR活性。实验和理论模拟研究表明,具有过渡金属无机纳米粒子的NCNTs显示出比纯CNTs更好的电化学性能[6-7]。尽管如此,这些材料的性能与商用Pt/C材料的性能还存在差距,限制了其在高能量密度器件中的应用。金属有机骨架(MOFs)被认为是制备不同多孔结构电催化剂最有吸引力的前驱体或模板之一[8-10]。例如,利用MOFs材料制备掺杂杂原子的纳米多孔碳[11]、碳包覆的金属[12]以及它们的复合纳米结构[13]。其中,锌基沸石咪唑盐骨架(ZIF8)是由2-甲基咪唑为有机配体,硝酸锌提供金属离子组成的有机框架结构,由于其富含C和N元素,所以是制备高性能氮掺杂碳材料的典型牺牲模板。然而MOFs材料本身导电性较差、高温热解后结构易坍塌且缺乏互连、石墨化程度低[4,13]等,使其在ORR反应中不能满足电子快速转移的要求,因此需要引入二次碳源(如氧化石墨烯,碳纳米管等)或过渡金属源调整其形貌来提高ORR活性。在氮掺杂碳过渡金属复合材料中,温度过高容易引起金属的聚集,不利于活性位点的形成。在ZIF8中,二甲基咪唑配体中有明确的锚定位点,它可以作为多位点锚定来分散金属,以抑制金属纳米粒子在热解过程中的聚集[14]。此外,锌的沸点为907 ℃[15],高温下由于锌的挥发,样品会形成丰富的纳米孔结构,有利于电子的传输。
本工作采用改良Hummers制备氧化石墨烯,以氧化石墨烯为载体,ZIF8为模板,尿素提供碳和氮源,无水FeCl3为金属源,通过高温热解,ZIF8分解,在氮掺杂石墨烯片上原位生长碳纳米管多孔复合结构,这种三维多孔结构有利于抑制金属的聚集,暴露更多的活性位点,促进电子的快速传输,有助于电催化性能的提高。
1 实验
1.1 催化剂的制备
本工作采用改良Hummers法进行催化剂的制备[16]。将5 g膨胀石墨粉和2.5 g NaNO3加入115 mL浓H2SO4中,在低于10 ℃的冰水浴中搅拌25 min,接着缓慢加入15 g KMnO4。25 min后升温至35 ℃,搅拌45 min后加入230 mL去离子水,升温至98 ℃,继续搅拌45 min。然后加入350 mL去离子水,待冷却至室温后加入30 mL H2O2,搅拌30 min,接着加入30 mL HCl,搅拌30 min,得到混合溶液。将混合溶液静置冷却后用去离子水离心洗涤pH值接近6,最后超声2 h,得到氧化石墨烯悬浮液(GO)。
将3 g尿素加入50 mL(1 mg/mL)氧化石墨烯悬浮液中,超声2 h,得到均匀的棕色溶液。-80 ℃中冷冻12 h得到棕色固体,再置于冷冻干燥机中干燥48 h,得到尿素与氧化石墨烯复合的棕色粉末(N-GO)。
将4 g二甲基咪唑溶于25 mL甲醇中,搅拌10 min,得到二甲基咪唑溶液。取1 g Zn(NO3)·6H2O溶于10 mL甲醇中,搅拌10 min,得到Zn(NO3)·6H2O溶液。将二甲基咪唑溶液缓慢滴加到Zn(NO3)·6H2O溶液中,得到均匀的乳白色溶液,室温下搅拌12 h,在8000 r/min转速下离心20 min后,用甲醇洗涤3次,并在60 ℃下干燥5 h,得到ZIF8样品。然后,取120 mg ZIF8,加入5 mL甲醇中,搅拌15 min,得到溶液A。60 mg的无水FeCl3加入5 mL甲醇,搅拌15 min,得到溶液B。将溶液A与溶液B混合搅拌12 h,在8000 r/min转速下离心20 min后,用甲醇洗涤3次,并在60 ℃下干燥5 h,得到Fe/ZIF8样品。
取1.25 g N-GO粉末与40 mg Fe/ZIF8,在研磨器中研磨混合30 min,在Ar气氛中以3 ℃/min升温到550 ℃,保温2 h,接着升温至900 ℃,保温2 h,得到N/GO和Fe-ZIF8的复合材料,命名为N-GO@Fe/ZIF8-900。此外,为了探究N-GO@Fe/ZIF8-900的结构组成,分别将40 mg的Fe和ZIF8替换成等量的无水FeCl3和ZIF8,相同条件下,在900 ℃煅烧得到N-GO@Fe-900,N-GO@ZIF8-900作为对照样品。
1.2 测试与表征
工作电极的制备:将5 mg催化剂溶于500 μL酒精中, 30 min超声后加入470 μL去离子水,超声30 min后加入30 μL Nafion溶液,最后超声30 min形成均匀的油墨状溶液。分别两次取10 μL油墨状溶液涂于玻碳电极上,形成直径5 mm的催化剂膜涂层,催化剂负载量为0.404 mg/cm2。
采用X射线粉末衍射仪(D8 ADVANCE)对样品进行物相分析,衍射源为CuKα(λ=0.15406 nm,扫描速率为3 (°)/min;拉曼光谱分析采用激光拉曼光谱仪(Horiba Lab RAM HR Evolution);采用扫描电子显微镜(FEI Quanta 450),透射电子显微镜(Tecnai G2 F30)观察分析样品的微观形貌;使用比表面积分析仪(Micromeritics ASAP2020)进行比表面积和孔结构分析;电化学性能测量在电化学工作站(PINE AF)上进行;电池性能在电池测试系统(CT2001A)和电化学工作站(CHI760E)上进行。利用三电极体系,饱和甘汞电极作为参比电极(换算为可逆氢电极(RHE)),铂片作为对电极,涂覆催化剂的旋转环盘电极作为工作电极,在分别通入饱和O2和N2的0.1 mol的氢氧化钾溶液中进行ORR和OER测试。ORR测试在0.2~1.2 V的电势窗口内进行,扫描速率为10 mV/s,电极旋转速率为400~2025 r/min。OER测试在1.2~2.0 V的电势窗口内进行,扫描速率为10 mV/s,电极旋转速率为1600 r/min。
锌空气电池的组装:将15 mg的CNTs和5 mg催化剂置于50 mL的乙醇中超声分散均匀,然后真空抽滤,形成一张均匀的膜,再用切片机切成直径为8 mm的圆片作为催化层。催化剂负载量为0.2 mg/cm2,阴极从里向外依次为聚四氟乙烯膜、催化层、离子交换膜和泡沫镍,清洁的锌板和6.0 mol/mL的KOH分别用作阳极和电解液,用相同的方法将5 mg的20%Pt/C(质量分数)催化剂与15 mg的CNTs组装成锌空气电池。
2 结果与讨论
2.1 催化剂的组成与结构
图1为N-GO@Fe/ZIF8-900样品的合成示意图。以石墨烯为基底,ZIF8和尿素提供碳和氮源,无水FeCl3为金属源,通过高温热解形成氮掺杂石墨烯片上原位生长碳纳米管结构。以氮掺杂石墨烯为基底,一方面提高导电性,另一方面碳氮形成C-N活性中心,同时ZIF8也为过渡金属Fe提供更多的锚定位点。在煅烧过程中,石墨烯片上的含氧基官能团与Fe/ZIF8中的金属离子有静电相互作用[17],将Fe/ZIF8锚定在氮掺杂氧化石墨烯片上,在高温下由于Fe的催化[18],ZIF8和N/GO原位生长出碳纳米管结构。最终碳纳米管与二维的石墨烯片形成氮掺杂石墨烯原位生长碳纳米管三维多孔结构。

图1 N-GO@Fe/ZIF8-900合成示意图Fig.1 Schematic diagram of N-GO@Fe/ZIF8-900 synthesis
图2为N-GO@ZIF8-900,N-GO@Fe-900,N-GO@Fe/ZIF8-900样品的SEM图。可知,煅烧后的ZIF8颗粒分布在氮掺杂石墨烯表面,未出现碳纳米管(图2(a));在石墨烯片上衍生出碳纳米管结构,Fe/Fe3O4被包覆在金属的顶端,直径为(710±5) nm(图2(b));在高温煅烧过程中,金属离子与氧化石墨烯片中的含氧官能团之间有着强相互作用,将Fe掺入ZIF8中形成Fe-ZIF8,并均匀锚定在GO纳米片上(图2(c))。以GO为基底,ZIF8为模板,尿素作为碳氮源,过渡金属源作为催化剂,催化碳纳米管生长[19-20],长成的碳管直径为(70±5) nm,仅为N-GO@Fe-900碳管直径的1/10左右。由于金属催化碳纳米管生长,碳纳米管的直径类似金属颗粒的直径。因此,通过将Fe-ZIF8与N/GO结合,衍生出直径较小的金属颗粒,有效抑制金属的聚集。

图2 N-GO@ZIF8-900(a),N-GO@Fe-900(b)和N-GO@Fe/ZIF8-900(c)的SEM图Fig.2 SEM images of N-GO@ZIF8-900(a),N-GO@Fe-900(b) and N-GO@Fe/ZIF8-900(c)
图3为N-GO@Fe/ZIF8-900样品的TEM图。可以看出,N/GO和Fe-ZIF8原位生长出碳纳米管结构,碳纳米管具有中空结构,含铁纳米颗粒(暗区)被包覆在碳纳米管的顶端。由氮掺杂碳纳米管(NCNTs)顶端的高分辨TEM放大图可知,间距为0.21 nm的晶格条纹为立方铁的(110)晶面,而间距为0.34 nm的晶格条纹为碳的(002)晶面,证实铁纳米粒子被限制在石墨碳纳米管中。由N-GO@ Fe/ZIF8-900样品中少量未分解完全的Fe/ZIF8透射电镜图可知,Fe/ZIF8主要以球形颗粒存在,含铁纳米颗粒显示晶格条纹间距0.21 nm,为立方铁(110)面,证明Fe成功掺入ZIF8框架中,形成氮掺杂石墨烯原位生长碳纳米管的三维多孔结构。Fe与N的掺入可以形成很多Fe-Nx活性位点[21],这种三维多孔结构不仅有利于反应物在催化过程中的扩散,也有利于暴露更多的活性位点,从而促进电子的传输。

图3 N-GO@Fe/ZIF8-900的TEM图Fig.3 TEM images of N-GO@Fe/ZIF8-900
2.2 XRD,Raman和比表面积测试分析
图4为N-GO@Fe-900,N-GO@Fe/ZIF8-900的XRD谱图,Raman谱图和比表面积测试图。图4(a)为N-GO@Fe-900,N-GO@Fe/ZIF8-900样品的XRD谱图,两者的衍射峰与Fe(PDF No.87-0721),Fe3O4(PDF No.79-0418)和C(PDF No.75-1621)保持一致,表明金属主要以Fe和Fe3O4的形式存在,碳主要以石墨碳的形式存在,说明氧化石墨烯在煅烧过程中被还原[22]。
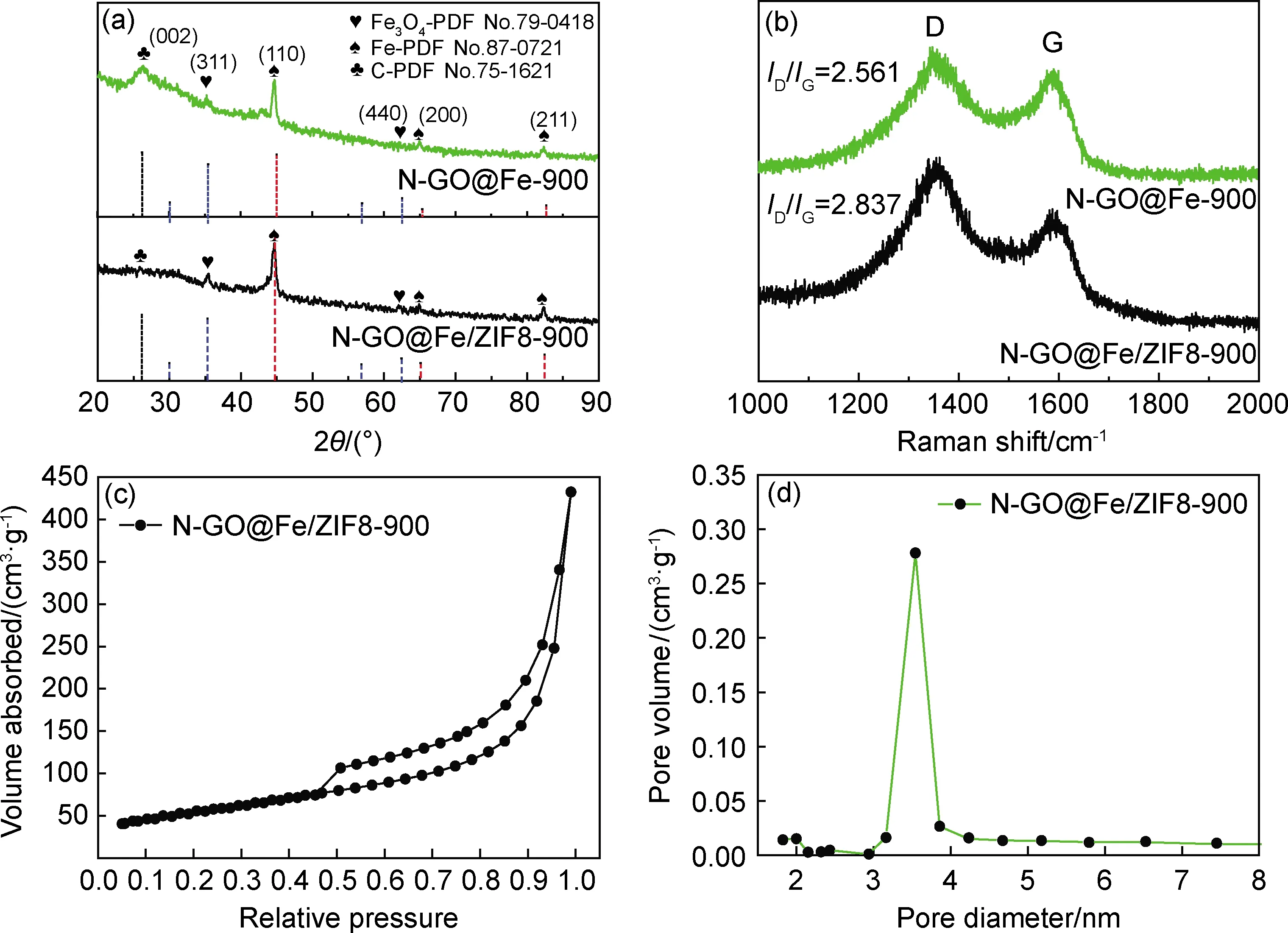
图4 N-GO@Fe-900,N-GO@Fe/ZIF8-900的XRD谱图(a)和Raman光谱图(b),N-GO@Fe/ZIF8-900的N2吸附/脱附等温曲线(c)和孔径分布曲线(d)Fig.4 XRD patterns(a) and Raman spectra(b) of N-GO@Fe-900 and N-GO@Fe/ZIF8-900,N2 adsorption/desorption isotherm(c) and pore size distribution curve(d) of N-GO@Fe/ZIF8-900
其中N-GO@Fe-900中Fe衍射峰的平均半高宽为0.427°,N-GO@ Fe/ZIF8-900中Fe衍射峰的平均半高宽为0.474°,峰宽较宽,说明晶粒尺寸较小,有利于活性位点的暴露[15,23],从而促进ORR/OER反应进程。图4(b)为N-GO@Fe-900,N-GO@Fe/ZIF8-900的拉曼光谱图,可以清楚地观察到1340 cm-1(D峰)和1595 cm-1(G峰)处的两个特征峰,其中D峰表示C原子晶格的缺陷,G峰表示C原子sp2杂化的面内伸缩振动。D峰和G峰的强度比为ID/IG,反映了碳材料的缺陷。相比于N-GO@Fe-900的ID/IG值(2.561),N-GO@Fe/ZIF8-900催化剂中ID/IG的值(2.837)较大,表明N-GO@Fe/ZIF8-900中存在更加丰富的缺陷位点,有利于反应过程中氧的吸附。通过N2吸附/脱附测试进一步表征900 ℃下N-GO@Fe/ZIF8样品的比表面积和多孔结构,如图4(c)所示,Ⅳ型等温线线和磁滞(P/P0>0.45)处闭合,证实N-GO@Fe/ZIF8中存在大量的介孔结构,其中N-GO@Fe/ZIF8-900样品的比表面积达到196.1 m2/g。图4(d)的孔径分布曲线显示,N-GO@Fe/ZIF8-900样品催化剂由介孔组成,孔径为3.53 nm,孔容达到0.67 cm3/g,表明在900 ℃下尿素的分解(NH3CO(NH2)2=2NH3↑+HNCO)及锌的挥发可以提供大的比表面积,有利于电子的传输。
2.3 XPS分析
采用X射线光电子谱(XPS)探究N-GO@Fe/ZIF8-900催化剂的成键和表面元素组成,如图5所示。图5(a)显示N-GO@Fe/ZIF8-900催化剂中C,N,O,Fe和Zn的原子分数分别为85.82%,1.56%,6.62%,5.89%和0.11%,Zn的含量相当低,表明在煅烧过程中Zn基本挥发完全。图5(b)为N-GO@Fe/ZIF8-900的C1s谱图,其中285.3 eV的特征峰对应碳氮键[17,24-25],表明氮原子成功掺入碳骨架中。
图5(c)显示N-GO@Fe/ZIF8-900的N1s谱图。N1s有四个特征峰,397.3 eV对应吡啶氮、398.2 eV对应吡咯氮、400.1 eV对应石墨氮、401.9 eV对应氧化氮,其中石墨氮和吡啶氮占总氮原子含量的百分比高达60.2%,如图5(e)所示。氮掺杂到石墨烯结构中会导致电子分布不均,这些变化将促进氧的吸附和O—O键的解离,边缘掺杂的石墨氮和吡啶氮降低了氧在相邻碳原子上吸附的能垒,加速氧还原中电子的转移,所以石墨氮和吡啶氮被认为是提高催化剂活性的主要基团[26]。图5(d)为N-GO@Fe/ZIF8-900的Fe2p XPS谱图,Fe2p有六个自旋轨道峰和一个卫星峰。Fe0对应718.9 eV和706.5 eV两个峰,710.1 eV和723.3 eV的两个峰被分配给Fe2+,Fe3+对应713 eV和732 eV的两个峰,意味着Fe和Fe3O4存在,与XRD测试结果相符。图5(e)中,N-GO@ Fe/ZIF8-900的Fe2p的Fe0∶Fe2+∶Fe3+的比例计算为10∶67∶23。丰富的Fe2+空位有利于形成Fe-Nx的活性中心[17],密度泛函理论计算证实Fe与N配位的相互结构有利于氧的吸附,Fe-Nx位点可以有效地裂解O—O键,在一定程度上提高ORR性能。此外,共存的Fe0,Fe2+和Fe3+氧化还原过程中电子的快速转移明显提高复合材料的导电性,供体-受体位点可以由铁阳离子的价态交替提供,可逆地吸附/脱附氧提高ORR/OER活性。

图5 N-GO@Fe/ZIF8-900的XPS全谱图(a),C1s(b),N1s(c),Fe2p谱图(d)及不同类型N和Fe原子掺杂条形图(e)Fig.5 XPS spectrum of N-GO@Fe/ZIF8-900(a),C1s spectra(b),N1s spectra(c),Fe2p spectra(d),and bar graph of different types of N and Fe atom doping(e)
2.4 ORR性能测试分析
图6为不同催化剂的ORR性能测试图。图6(a)为不同催化剂样品的LSV曲线,N-GO@Fe/ZIF8-900的半波电位达到0.885 V,优于N-GO@ZIF8-900(0.821 V),N-GO@Fe(0.849 V)和Pt/C(0.856 V),显示出最优的氧还原性能。图6(b)为N-GO@Fe/ZIF8-900在不同转速下的LSV曲线,电流密度随着转速的增加而均匀增加。图6(c)显示N-GO@Fe/ZIF8-900催化剂样品在不同电势下利用K-L[15]方程计算获得的氧还原反应过程中的电子转移数n,当电势在0.2~0.5 V时,催化剂N-GO@Fe/ZIF8-900的电子转移数在3.67~3.98之间,表明在ORR过程中的电子转移符合4e-转移。图6(d)为N-GO@Fe/ZIF8-900催化剂样品在O2中稳定性测试曲线,可见持续运行16000 s后的电流密度仍保持在初始电流密度的87.6%左右,而Pt/C的电流密度保留率仅为70.6%,表明在碱性溶液中催化剂N-GO@Fe/ZIF8-900的稳定性优于商用Pt/C。

图6 各催化剂在通入饱和O2的0.1 mol KOH溶液下的LSV曲线(转速为1600 r/min,扫描速率为5 mV/s) (a),N-GO@Fe/ZIF8-900催化剂不同转速下的LSV曲线(b),K-L曲线(c),I-T测试曲线(d)Fig.6 LSV curves of different catalysts in 0.1 mol KOH saturated with O2 (rotating speed:1600 r/min,scanning rate: 5 mV/s)(a),LSV curves(b),K-L curves(c),I-T test curves(d) of N-GO@Fe/ZIF8-900 catalyst under different speeds
2.5 OER性能测试分析
图7为N-GO@Fe/ZIF8-900,Pt/C及IrO2的氧析出性能测试图。图7(a)的LSV曲线显示,在10 mA/cm2的电流密度下N-GO@Fe/ZIF8-900样品对应电位为1.811 V,优于贵金属Pt/C (1.968 V),与IrO2(1.75 V)性能相当。图7(b)为N-GO@Fe/ZIF8-900,Pt/C及IrO2的Tafel曲线,可以看出N-GO@Fe/ZIF8-900样品的Tafel斜率为129.1 mV/dec,优于Pt/C(278.3 mV/dec),表明N-GO@Fe/ZIF8-900具有优异的双功能催化活性。

图7 N-GO@Fe/ZIF8-900,Pt/C及IrO2样品的LSV曲线(a)及Tafel曲线(b)Fig.7 LSV curves(a) and Tafel curves(b) of N-GO@Fe/ZIF8-900,Pt/C and IrO2 samples
2.6 电池性能测试分析
图8为N-GO@Fe/ZIF8-900与Pt/C组装成锌空气电池后的性能测试图。图8(a)为组装成的锌空气电池在5 mA/m2条件下的循环稳定性测试对比图。起始时N-GO@Fe/ZIF8-900样品的电压间隙(充电电压-放电电压)为0.949 V,而贵金属Pt/C催化剂的电压间隙为0.932 V。经过130 h的测试,两者的电压间隙分别为0.961,1.004 V,N-GO@Fe/ZIF8-900样品的电压间隙仅增加0.012 V,表明N-GO@Fe/ZIF8-900样品具有更加优异的稳定性。图8(b)为电流密度为10 mA/cm2时N-GO@Fe/ZIF8-900和Pt/C的比能量曲线。可以看出,N-GO@Fe/ZIF8-900的比能量达到886.2 mW·h·g-1,远大于贵金属催化剂Pt/C的比能量(791.04 mW·h·g-1)。图8(c)为N-GO@Fe/ZIF8-900和Pt/C的极化曲线。N-GO@Fe/ZIF8-900的充放电电压间隙小于Pt/C的电压间隙,表明N-GO@Fe/ZIF8-900样品组装成锌空气电池时,具有优于Pt/C的充放电性能。图8(d)为N-GO@Fe/ZIF8-900和Pt/C的功率密度曲线。N-GO@Fe/ZIF8-900的功率密度达到73.44 mW/cm2,优于贵金属催化剂Pt/C的功率密度(57.12 mW/cm2),表明N-GO@ Fe/ZIF8-900样品组装的锌空气电池具有优异的电化学性能。

图8 电流密度为5 mA/m2时组装锌空气电池的循环稳定性测试曲线(a),电流密度为10 mA/cm2 时的比能量曲线(b),充放电极化曲线(c)及功率密度曲线(d)Fig.8 Cycle stability test curves of assembled zinc air battery at 5 mA/m2(a),specific energy curves at 10 mA/cm2(b),charge and discharge polarization curves(c) and power density curves(d)
3 结论
(1)通过直接热解法构建一种氮掺杂石墨烯片上原位生长碳纳米管多孔复合结构。N-GO@Fe/ZIF8-900催化剂与N-GO@Fe-900相比,有效抑制金属的聚集。
(2)复合催化剂N-GO@Fe/ZIF8-900表现出良好的ORR/OER性能,在催化氧还原过程中,半波电位达到0.885 V,优于Pt/C(0.856 V)。在催化氧析出过程中,10 mA/cm2的电流密度下N-GO@Fe/ZIF8-900样品对应电位为1.811 V,优于贵金属Pt/C(1.968 V),与IrO2(1.75 V)性能相当,表明N-GO@Fe/ZIF8-900具有良好的双功能催化活性。
(3)复合催化剂N-GO@Fe/ZIF8-900组装成锌空气电池,具有优于Pt/C的循环稳定性,比能量和功率密度分别达到886.2 mW·h·g-1和73.44 mW/cm2,高于贵金属Pt/C的比能量(791.04 mW·h·g-1)和功率密度(57.12 mW/cm2),有望成为在可充电锌空气电池中贵金属催化剂的替代品。
猜你喜欢
分子催化(2022年1期)2022-11-02
昆钢科技(2022年1期)2022-04-19
建材发展导向(2021年16期)2021-10-12
食品安全导刊(2021年20期)2021-08-30
纺织科学研究(2021年7期)2021-08-14
化工设计通讯(2021年5期)2021-05-26
军事文摘(2020年20期)2020-11-16
浙江农林大学学报(2020年2期)2020-04-22
物理化学学报(2020年1期)2020-04-02
智富时代(2018年3期)2018-06-11
