
对照品及流动相对海藻酸钠分子量测定的影响
2022-05-20 08:12张庆龙
青岛大学学报(自然科学版) 2022年2期
徐 蕾,王 瑾,陈 涛,张庆龙,杨 钊
(1.青岛大学 a.药学院,b.附属医院,青岛 266021;2.青岛市食品药品检验研究院,青岛 266071)
海藻酸钠作为一种海洋植物产物,是从褐色海藻植物中用稀碱提取的海藻酸的钠盐[1-2],因其良好的生物相容性和生物可降解性,在食品、生物医药、药物传递系统等领域具有广泛的应用前景[3-9]。海藻酸钠作为高聚糖醛酸,具有分子大小不均一的特点,其分子量及分布是控制质量的关键指标之一。一种海藻酸钠的分子量通常代表该组所有分子的平均值,表示方式有重均分子量(Mw)、数均分子量(Mn)。常用Mw/Mn表示分子量的分布,数值越大,表示分子量分布越宽;数值越小,表示分子量分布越窄。高效凝胶渗透色谱法是测定分子量及其分布最常用的方法,分离原理为凝胶色谱柱的分子筛机制。当药物分子进入色谱柱后,分子量较大的部分保留时间较短,而分子量相对较小的保留时间较长,按分子大小依次被洗脱,从而达到分离效果[10-14]。该方法适用范围广、便捷和重现性好,被常用于测定大分子物质的分子量及其分布。本文拟采用高效凝胶渗透色谱法比较分析不同厂家生产的9批海藻酸钠原料药的分子量及其分布[15-17],研究不同组分的对照品、不同组成的流动相对海藻酸钠分子量及其分布测定的影响,以期完善其质量标准,提升质量控制。
1 仪器与试药
1.1 仪器
Agilent1100高效液相色谱仪(配备G1362A示差折光检测器,Agilent公司);TSK gel G4000PWxl色谱柱(7.8×300 mm,TOSOH公司);电子天平(BT125D型,Sartorius公司);电子天平(BSA224S型,Sartorius公司);Agilent GPC数据分析软件(Agilent公司)。
1.2 试药
右旋糖酐分子量标准(套)对照品,中国食品药品检定研究院,批号 140637~646-201203;普鲁兰分子量标准(套)对照品,昭和电工株式会社,批号 190801。无水硫酸钠(分析纯,天津市科密欧化学试剂开发中心),叠氮钠(分析纯,国药集团化学试剂有限公司)超纯水由Millipore超纯水系统制备。
1.3 样品
A厂家6批样品批号分别为:391812002、391902001、391812004、391805006、391902002、391902003,编号1~6;B厂家3批样品批号分别为:LFR5/60、CR8133、CR8223,编号7~9。
2 方法
2.1 色谱条件
色谱柱:TSK gel G4000PWxl;柱温40 ℃;流速0.5 mL·min-1;进样量20 μL。检测器:示差折光检测器;检测池温度45 ℃。
2.2 溶液制备
2.2.1 供试品溶液 称取海藻酸钠样品0.1 g,用流动相溶解并定容至100 mL,滤过,取续滤液作为供试品溶液。
2.2.2 对照品溶液1 取已知分子量的右旋糖酐对照品适量,用流动相溶解并稀释至每1 mL中各约含1 mg的溶液,取续滤液即得。
2.2.3 对照品溶液2 取已知分子量的普鲁兰对照品适量,用流动相溶解并稀释至每1 mL中各约含1mg的溶液,取续滤液即得。
2.2.4 流动相 称取无水硫酸钠28.41 g,用水定容至2 000 mL,得0.1 mol·L-1的硫酸钠溶液,配置流动相A。流动相B为超纯水。
2.3 标准曲线的制作
取对照品溶液1和2各20 μL注入液相色谱仪,记录洗脱峰的保留时间。以对照品分子量的对数值为纵坐标、相应色谱峰的保留时间为横坐标,用GPC软件进行三阶线性拟合,得到线性回归方程。
2.4 对照品对分子量测定的影响
分别取对照品溶液1和2、供试品溶液,以流动相A进行测定,采用GPC软件计算,比较选用不同对照品的测定结果。
2.5 流动相对分子量测定的影响
取对照品溶液1和供试品溶液,分别以流动相A、流动相B进行测定,采用GPC软件计算,比较选用不同流动相的测定结果。
3 结果与讨论
3.1 试验方法学结果
3.1.1 系统适用性 取空白溶剂(0.1 mol·L-1硫酸钠溶液)与7号样品分别注入液相色谱仪,图1为7号样品出峰的色谱图,溶剂峰不干扰本品主峰。
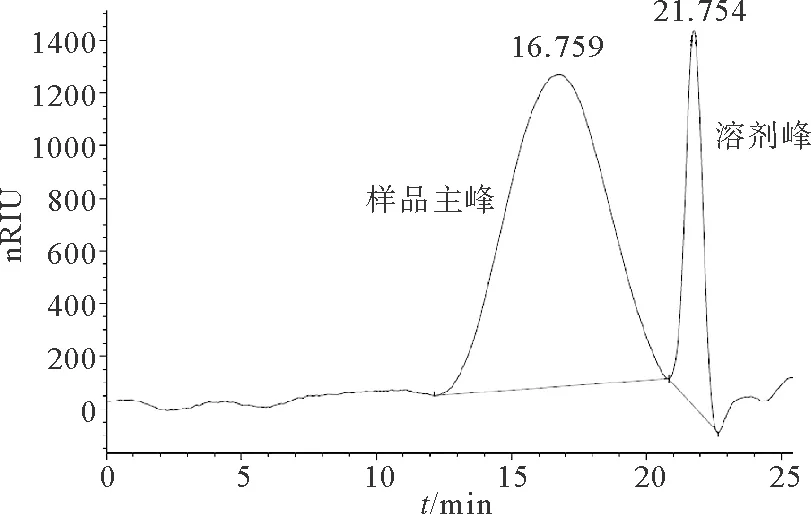
图1 7号样品色谱图
3.1.2 保留时间 右旋糖酐和普鲁兰多糖对照品各分子量的保留时间分别见表1、表2。测得供试品溶液主峰保留时间既在右旋糖酐对照品D1和D10主峰保留时间范围内,也在普鲁兰多糖对照品D1和D5主峰保留时间范围内。
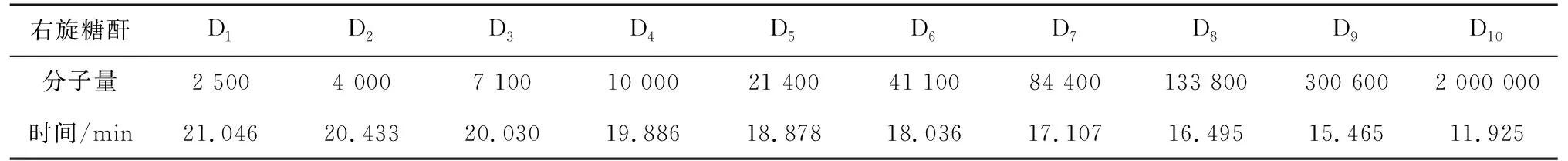
表1 右旋糖酐对照品洗脱时间

表2 普鲁兰多糖对照品洗脱时间
3.2 不同流动相对分子量测定结果的影响
实验选用多糖分子量测定常用的两种流动相纯水和硫酸钠溶液,分别考察两者对测定海藻酸钠分子量结果的影响。8号样品在不同流动相下的色谱图如图2所示,当以纯水为流动相时,色谱图形状异常;以硫酸钠溶液为流动相时,海藻酸钠的色谱峰峰型对称性较好,对保留时间影响较小。海藻酸钠在不同流动相下产生的色谱行为有如此大的差别,原因可能与海藻酸钠的多聚物结构有关,海藻酸钠独特的空间结构使其分子在不一样的溶液系统中会产生不同的聚集形态,藻酸盐分子和洗脱液之间的互相作用也可能导致保留时间发生变化。以纯水为流动相时,褐藻胶在水中产生阴离子排斥作用,分子处于伸展状态,流体力学体积较大,分离效果不稳定,出峰时间早。以强电解质溶液为流动相时,褐藻胶分子伸展减弱,色谱行为较稳定[18-19]。海藻酸钠的重均分子量(Mw)、数均分子量(Mn)和分子量分布宽度(D)的测定结果如表3所示。可以看出,Mw差距最小的是4号样品相差90 000;最大的是7号样品相差达到1 200 000;Mn差距最小的是1号样品相差50 000;最大的是7号样品相差达到1 100 000。样品的色谱峰会影响保留时间,表现为分布范围变宽或变窄,从而导致相对分子量降低或升高。
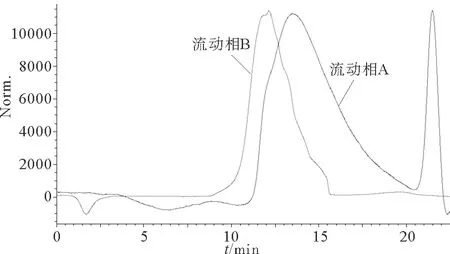
图2 8号样品色谱图
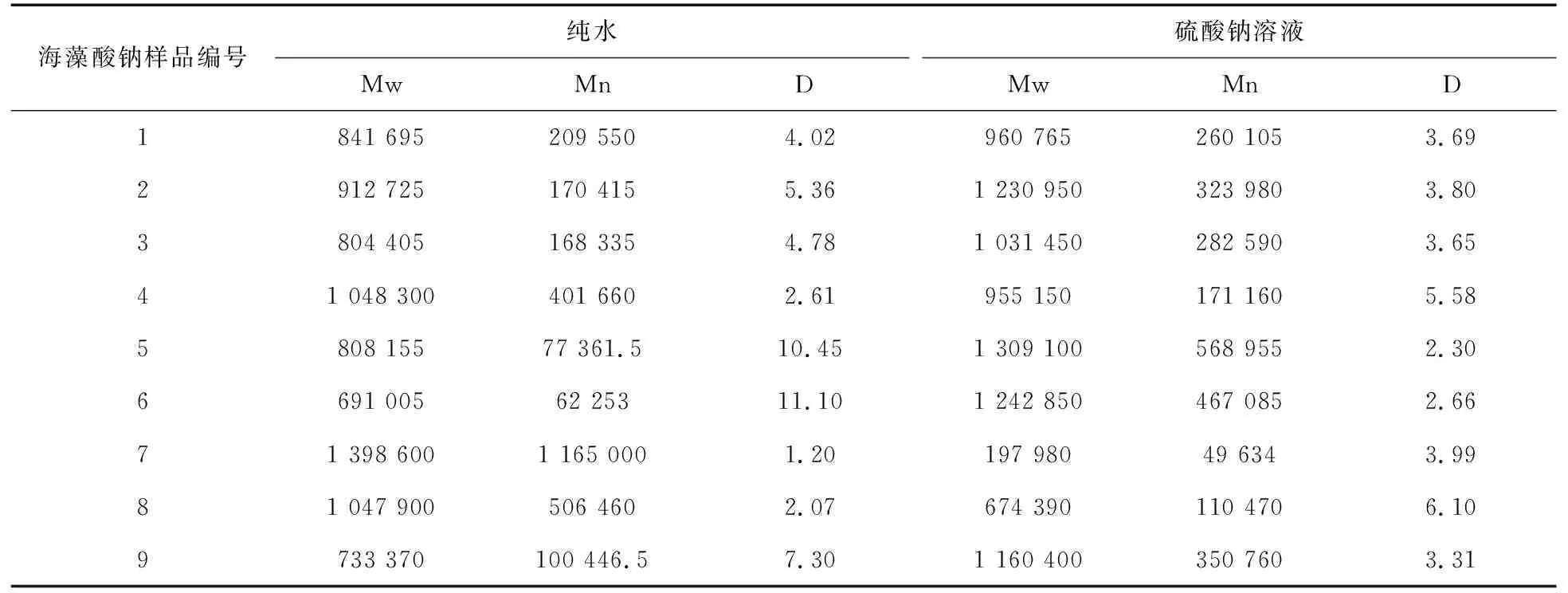
表3 不同流动相测定分子量与分子量分布结果比较
3.3 不同对照品对分子量测定结果的影响
使用同一根TSK G4000PWxl色谱柱,分别使用两种对照品进行检测,得右旋糖酐线性回归方程:Y=0.0009673888X3-0.08130449X2+0.5020784X+5.998968;普鲁兰线性回归方程:Y=-0.01530996X3+0.3353058X2-3.017180X+15.39076。采用GPC软件对海藻酸钠重均分子量(Mw)、数均分子量(Mn)和分子量分布宽度(D)的分析结果如表4所示。可以看出,在相同条件下,分别使用右旋糖酐和普鲁兰对照品,得到的相对分子量结果差距比较大;Mw测定结果偏差最小的是7号样品相差40 000;最大的是6号样品达到1 000 000。使用普鲁兰对照品制作标准曲线测得的样品分子量结果相对偏小,分子量分布D值也较使用右旋糖酐对照品制作标准曲线测得的结果低。随着Mw的变大,两种对照品测得的重均分子量差距也越来越大。HPGPC法在测定多糖分子量时需要适合的对照品,对照品的分子量分布情况越接近于样品,分子量测定值与真实值越接近。右旋糖酐及普鲁兰是适用于各种水溶性校正曲线的标准样品[20],也是目前广泛使用的分子量对照品。由于分子排阻色谱中的分子筛作用是使溶质分子按分子的大小顺序而非直接以分子量大小顺序洗脱,因此不同对照品的选择会直接影响其测定结果。
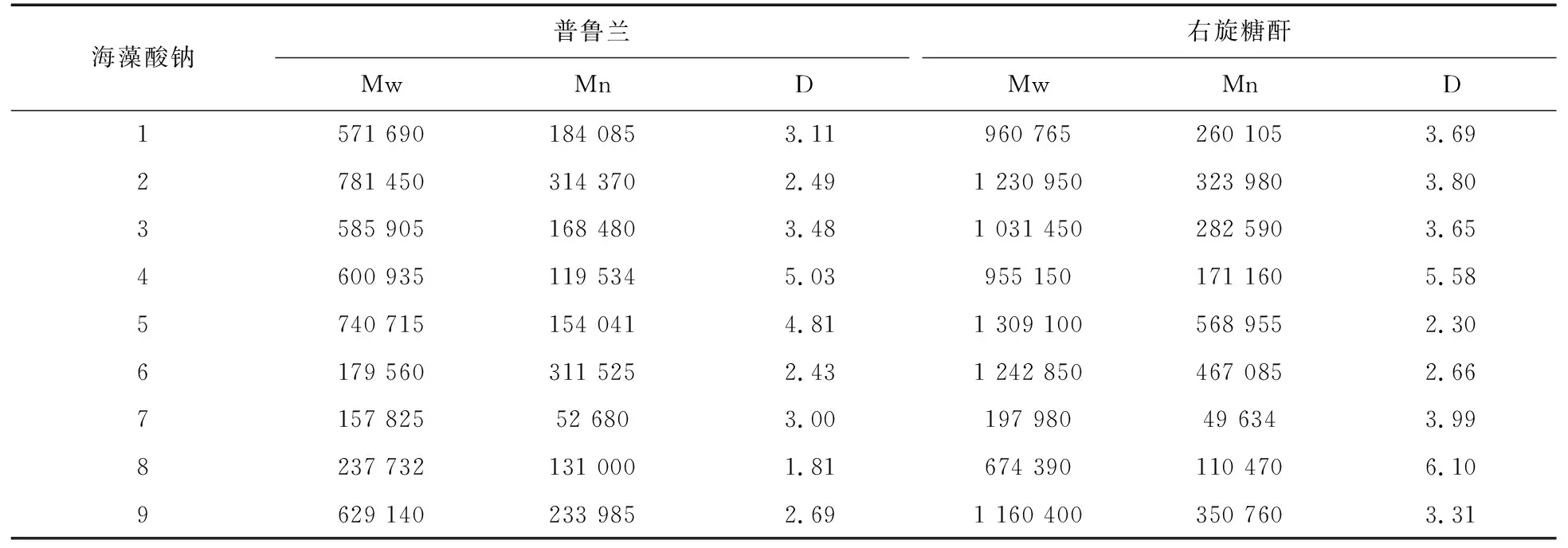
表4 不同对照品测定分子量与分子量分布结果比较
4 结论
实验发现在海藻酸钠分子量的测定中,对照品、流动相等色谱条件的变化会对不同样品的分子量及其分布趋势产生一定影响。结果表明分子量级小的样品可以选择普鲁兰为对照品,分子量级大的样品可以选择右旋糖酐为对照品;而硫酸钠溶液则是测定海藻酸钠分子量时较为理想的流动相选择。目前海藻酸钠的分子量测定没有统一质量标准。因此建议在其测定过程中,应充分注意不同色谱条件对测定结果的影响,选择合适的对照品、流动相,降低试验误差对结果的影响。
猜你喜欢
健康体检与管理(2022年4期)2022-05-13
作文周刊·小学四年级版(2022年8期)2022-03-11
保健与生活(2021年7期)2021-04-19
医学前沿(2021年18期)2021-04-14
海峡科技与产业(2019年4期)2019-10-26
家庭百事通·健康一点通(2018年9期)2018-10-12
食品与生活(2017年12期)2018-01-09
分析化学(2017年4期)2017-04-14
中国民族民间医药·下半月(2017年2期)2017-03-20
世界热带农业信息(2014年6期)2014-09-12
