
钒微合金钢中α-Fe/V4C3界面结构与稳定性的第一性原理计算
2022-05-19 05:07:08刘佳敏李林鲜温希平刘振宇王国栋
材料工程 2022年5期
唐 帅,刘佳敏,李林鲜,温希平,彭 庆,刘振宇,王国栋
(1 东北大学 轧制技术及连轧自动化国家重点实验室,沈阳 110819;2 法赫德国王石油矿产大学 物理系,沙特阿拉伯 达兰 31261)
随着不断增大的资源、能源和环境压力,绿色环保和轻量节能已成为汽车工业的重要发展趋势。研究表明,整车质量下降10%,油耗降低6%,CO2等排放量下降4%[1]。汽车车体约70%由钢铁材料构成,因此,开发应用高强度低合金钢(high strength low alloy steel,HSLA)是降低汽车自重的有效途径之一[2]。经过数十年的探索和实验,在钢中添加Cu,Cr,Ni,Mo等合金元素和V,Ti,Nb等微合金元素可以促进奥氏体(γ-Fe)再结晶,在热加工过程中析出TiC,NbC和VC等碳化物,钉扎晶界抑制晶粒长大,大幅改善钢板的韧性,获得强韧性匹配优良的钢材。同时,这些纳米级碳化物也可在铁素体(α-Fe)基体上析出,通过成分和工艺优化,在α-Fe析出尺寸约3 nm的碳化物,抗拉强度在805 MPa时,扩孔率仍可高达100%,在强化基体的同时仍可以保持良好的延展性和可成形性,被广泛应用于汽车零件的制造[3-5]。如何提高高温蠕变性能是耐热钢的重要研究内容,通过引入VC和NbC等碳化物可在高温抑制α-Fe的晶粒粗化,改善铁素体耐热钢的高温蠕变性能[6]。定量研究α-Fe基体与碳化物的界面特性参数是调控碳化物析出的重要手段,目前很难采用实验方法表征,而采用密度泛函理论的第一性原理计算可以定量研究α-Fe/碳化物的界面特性[7-8]。针对过渡金属化学计量比碳化物与铁基体的界面性质已有大量学者进行研究,结果表明,掺杂V,Nb,Mo,W等元素可降低复杂碳化物的界面能从而促进碳化物的形核析出[9-13],并且α-Fe/MC(M=V,Nb和Ta)界面结构稳定性与界面处Fe,M和C原子之间键能密切相关[10],碳化物的C原子与过渡金属(例如Ti,V和Zr)原子在界面处形成强烈的化学键[14],有利于提高界面稳定性,促进共格析出物的形核。但是对于过渡金属非化学计量比碳化物与α-Fe的界面研究相对较少。
由于V与C原子的结合能力较弱,在高强度钢中经常形成非化学计量比型V4C3[15],其具有高硬度、高熔点、耐磨耐蚀以及优良的导电、导热特性[16-17]。因此,研究α-Fe/V4C3的界面性质对于调控高强钢的析出行为具有重要的理论意义和实用价值。电子结构解析能够从电子相互作用角度了解金属/碳化物及其界面的稳定性[18],已广泛应用到Fe/TiC[19],Fe/NbC[20],Fe/WC[21]等不同界面的电子密度、电荷转移机制以及界面成键特征等方面。但是,目前对于α-Fe/V4C3界面的稳定构型以及电子结构的研究较少。因此,本工作通过第一性原理计算α-Fe/V4C3的三种界面构型的电子结构,用界面分离功分析最稳定的界面构型。从局域电荷密度、差分电荷密度和态密度方面解释原子间的微观作用机理,揭示α-Fe/V4C3形成稳定界面的本质原因。
1 计算方法
采用基于密度泛函理论,结合平面波赝势方法的VASP[22]软件包进行第一性原理计算。离子和电子的相互作用采用投影缀加平面波(projector augmented wave,PAW)[23],交换关联能采用Perdew等[24]基于PBE(Perdew-Burke-Ernzerhof)参数构建的PAW-PBE势。用PBE形式的广义梯度近似(generalized gradient approximation,GGA)描述交换相关泛函[25]。采用Broyden-Fleccher-Goldfarb-Shanno算法[26]完成几何优化以实现原子的充分弛豫。Fe,V和C选用的价电子组态分别为3p63d64s2,3p63d34s2和2s22p2。体相α-Fe和V4C3截断能为550 eV,k点采用Gamma方法取值为13×13×13,结构优化过程中,允许原子位置和晶格矢量充分弛豫,收敛精度为1×10-5eV/atom,每个原子的受力均小于0.1 eV/nm。为了得到精确的计算结果,首先对各晶胞进行结构优化,然后基于优化后的稳定晶体构建界面模型进行计算。界面由5层α-Fe和5层V4C3构成,由于含有磁性,因此界面考虑自旋极化,截断能为350 eV,k点取值为11×11×1,计算中只允许原子位置弛豫,收敛精度为1×10-4eV/atom,每个原子的受力均小于0.5 eV/nm。
2 结果与分析
2.1 体相α-Fe和V4C3的结构和性能
HLSA钢种基体为α-Fe,计算中采用体心立方(body-centre-cube,BCC)晶体结构,对α-Fe和V4C3的晶格常数进行优化计算,使用PAW-PBE方法得到的计算结果如表1所示[27-34]。可知,优化后的晶格常数与以往的文献计算值以及实验结果相一致,证明所使用的计算方法的正确性。

表1 V4C3和α-Fe优化后的晶格参数
2.2 界面几何结构
已知α-Fe与过渡金属碳化物之间的界面一般遵循Baker-Nutting取向关系,即(100)MC∥(100)α-Fe,[010]MC∥[011]α-Fe。对于界面模型,由于C空位的存在,V4C3与α-Fe接触的面(即终止面)有两种原子构型,一种是VC面,其中V原子和C原子的数目相等;另一种是V2C面,其中V原子的数目是C原子的2倍(见图1)。研究表明,终止面为V2C面时,界面能量低,稳定性更好[29],因此后续计算的界面结构模型的终止面均为V2C面。

图1 V4C3晶胞示意图
使用5层Fe原子和5层V4C3原子组成三种高度对称但不同原子堆积序列的(100)α-Fe/(100)V4C3界面结构进行晶格优化计算,其中Fe原子位于C原子之上的界面结构指定为“Fe-on-C”构型;Fe原子位于V原子之上的界面结构指定为“Fe-on-V”构型;Fe原子有2个C原子和2个V原子作为最近邻原子的界面结构指定为“Bridge”构型,如图2所示。

图2 三种不同原子堆积序列的(100)α-Fe/(100)V4C3界面构型
2.3 界面分离功
界面分离功Wsep是将界面分离成2个自由表面所需的能量[8],利用界面分离功可研究不同合金元素在(100)α-Fe/(100)V4C3界面处的稳定性和界面原子的结合强弱,用于讨论界面的能量稳定性。界面分离功可以通过界面与结合前的2个自由表面之间总能量的差异来计算,如式(1)所示。
(1)
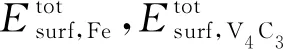
不同构型的Wsep如表2所示。原子弛豫后,Wsep的大小顺序为Fe-on-C>Bridge>Fe-on-V。Wsep最大的是Fe-on-C界面结构,表明Fe-on-C原子构型界面的稳定性最强,其次是Bridge构型,最后是Fe-on-V构型。Fe-on-C型α-Fe/V4C3的Wsep为4.29 eV,与Chen等[35]的计算结果(3.68 eV)略有差异,其原因是本工作考虑了C空位的影响。α-Fe/TiC与α-Fe/VC的Wsep相差不大,但都小于α-Fe/V4C3的Wsep,可见α-Fe/V4C3界面的稳定性更好。
界面距离d0反映原子间的吸引或者排斥作用,可用来描述界面原子间的结合能力(见表2)。通过测试,α-Fe/V4C3弛豫之前的d0为0.19 nm,晶胞的总能量最低,结构最稳定,因此选取0.19 nm为最佳界面距离,以求出经过弛豫后的平衡界面距离。原子弛豫后α-Fe/V4C3界面距离顺序为Fe-on-V>Bridge>Fe-on-C。比较三种结构,Fe-on-C构型d0降低,而Fe-on-V以及Bridge结构d0增大,表明Fe-on-C结构在原子优化过程中Fe和C原子彼此吸引,在界面处形成较强的化学键,从而促使d0降低,而Fe和V原子之间的键合作用弱,原子优化导致界面处化学键的削弱或断裂,引起d0增大,界面稳定性降低,进一步说明Fe-on-C 构型的界面原子之间具有更强的化学键键合。

表2 α-Fe/V4C3三种界面构型弛豫前后的界面距离和分离功
2.4 电子结构
界面成键特征对界面的性质影响很大,界面原子之间的结合取决于界面电子结构,可用电荷密度、差分电荷密度和态密度[36]描述。为了更深入地了解界面处的电子相互作用和电荷分布,计算α-Fe/V4C3三种界面构型的界面电子局域化函数(electron localization function,ELF)、差分电荷密度和态密度(density of states,DOS),以此判断电荷转移情况和成键特征。
图3为三种界面构型沿(100)平面电子局域化函数和差分电荷密度图。实空间中不同位置的电子定域程度可以通过ELF来呈现,其数值范围一般为0~1。ELF值越高,电子的定域性越高,说明域内的电子被限制在这个区域内的程度越高;ELF值越低,电子的定域性越低,表明电子的定域能力弱。图3(a-1)中,Fe-on-C界面处的C原子附近形成了高电荷密度带。图3(c-1)中,在结构弛豫之前Bridge结构中Fe原子距离最近邻C和V原子是相等的,结构弛豫后,Fe原子与最近邻C的距离明显小于与V原子的距离,Fe—C键长缩短。图3(b-1)中,Fe-on-V结构界面处Fe和V原子间电荷密度较低,表明Fe和V原子的键合作用较弱,以上结果说明Fe—C形成的化学键强于Fe—V。

图3 弛豫后α-Fe/V4C3三种界面构型(100)面的电子局域化函数(1)和差分电荷密度(2)
差分电荷密度是原子成键前后的电荷密度之差,差分电荷密度图可以直观地看出各个原子的成键情况(如电子得失、成键的极性强弱以及通过电荷的分布形状来判断成键的轨道)。原子弛豫后,三种界面的差分电荷密度如图3(a-2),(b-2),(c-2)所示。可知界面处电荷转移差异较大,呈局域化特征,这意味着电荷重新分布主要发生在界面或者附近。Fe-on-C界面的差分电荷密度如图3(a-2)所示,界面Fe侧失去更多的电荷,导致界面处Fe原子存在电荷贫化区。而损失的部分电荷转移给最近邻的C原子,界面呈局部离子性特征。由于C原子具有很强的电负性,易得到电子,界面处的电子向C原子侧移动,因此界面上同时存在电荷耗尽和累积区域,形成很强的极性共价键。Fe-on-V界面的差分电荷密度如图3(b-2)所示,靠近界面的V原子存在电荷耗尽区,丢失的电荷也会转移到界面,与Fe原子的电荷混合累积,因此界面形成混合离子/共价键。由于Bridge结构中Fe原子不在(100)面上,所以α-Fe的差分电荷分布不明显,如图3(c-2)所示。由差分电荷密度可推测出界面Fe侧均存在电荷耗尽区,即Fe原子失去电荷,转移至界面区域,从而促进界面原子成键。Fe-on-C与Fe-on-V界面的化学键均为混合离子/共价键,Fe—C原子之间的原子相互作用更强,因此Fe-on-C界面结构更稳定。
为了进一步理解Fe—C成键特征,图4分别给出了α-Fe/V4C3三种界面结构的总态密度(total density of states,TDOS)和Fe-d,V-d,C-p轨道的分波态密度(projected density of states,PDOS)。可知,三种构型的态密度相差较小,费米能级处(EF)的电子不为零,界面呈现明显的金属特征,费米能级两侧有尖峰,存在较宽的赝能隙,表明界面的稳定性较好。Fe-on-C,Fe-on-V和Bridge三种构型在费米能级处的态密度分别为7.074,8.306 states/eV和10.086 states/eV。Fe-on-C的值最小,表明该构型更加稳定。由PDOS可知,在Fe-on-V和Bridge构型中,Fe-d,V-d和C-p轨道在费米能级以下不存在明显的杂化峰,说明Fe原子与C原子和V原子之间相互作用较弱,这也是两种构型界面分离功较小的原因。而对于Fe-on-C构型,在-4.5~-2.5 eV时,Fe-d和C-p轨道具有明显的共振峰,说明此处发生电子轨道杂化,形成Fe—C共价键,这与电荷密度分析结构一致。并且发现三种构型界面处的C原子和V原子的PDOS皆有程度不同的不对称分布特征,说明由于与界面处Fe原子的杂化作用,使得界面处的C原子和V原子呈现磁性特征,特别是在Fe-on-V构型的V原子中,由于其与Fe原子距离过近,从而呈现较强的磁性。

图4 α-Fe/V4C3三种界面构型的总态密度和分波态密度
3 结论
(1)Fe-on-C构型的界面分离功最大,原子弛豫后界面距离减小,界面稳定性最强。
(2)Fe-on-C构型中界面Fe原子存在电荷贫化区,失去的电荷转移给界面处C原子,在界面处形成较强的混合离子/共价键。
(3)Fe-d轨道与C-p轨道在-4.5~-2.5 eV的区域内发生电子轨道杂化,形成Fe—C共价键。
猜你喜欢
高中数理化(2023年6期)2023-08-26 13:28:24
辽宁科技大学学报(2022年5期)2023-01-04 12:45:34
上海金属(2022年6期)2022-11-25 12:24:20
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08 00:48:08
原子与分子物理学报(2020年5期)2020-03-17 06:59:34
模具制造(2019年3期)2019-06-06 02:11:04
考试周刊(2018年39期)2018-04-19 10:39:44
北京航空航天大学学报(2017年10期)2017-04-20 08:51:23
上海金属(2016年4期)2016-11-23 05:38:50
航天返回与遥感(2014年4期)2014-07-31 17:47:47
