
复合益生菌产品菌种鉴定及活菌定量检测方法
2022-05-17 13:48周立光杨明喆冯会粉刘艺茹刘蕊张欣葛媛媛张旭光刘佳奇程坤于学健姚粟
食品与发酵工业 2022年9期
周立光,杨明喆,冯会粉,刘艺茹,刘蕊,张欣,葛媛媛, 张旭光,刘佳奇,程坤,于学健,姚粟*
1(中国食品发酵工业研究院有限公司,北京,100015)2(汤臣倍健有限公司,广东 广州,510663)
益生菌是一类活的微生物,当摄取足够数量时,对宿主健康有益[1],已广泛应用于食品、药品、膳食补充剂、饲料等领域[2]。益生菌的功能具备菌株特异性[3],且需要摄入足够的数量才能达到预期功效,因此在选择益生菌产品时,其活菌数量尤为重要。《益生菌类保健食品申报与审评规定(征求意见稿)》规定“益生菌类保健食品在其保质期内活菌数目不得少于106CFU/mL(g)”。近年来,随着益生菌产业的快速发展,多元化的复合益生菌产品不断涌现,对复合益生菌产品的菌种水平进行精确鉴定并确认其活菌数是确保产品质量的关键。
目前,乳酸菌的检测主要基于传统培养法,通过使用选择性培养基分离益生菌产品中所含的微生物[4],进而通过BIOLOG[5]、FT-IR以及MALDI-TOF[6]等表型方法和16S rRNA基因[7]、功能基因[8]等基于分子标记基因的遗传学手段来进行微生物菌种鉴定。尽管基于全基因组测序(whole genome sequencing,WGS)技术的平均核苷酸一致性(average nucleotide identity,ANI)[9]和单核苷酸多态性(single nucleotide polymorphism,SNP)分析[10]技术可以实现物种的精确鉴定,但仍需要获得纯培养物。基于16S rRNA基因序列的高通量测序,可以揭示产品属水平的物种组成,并能检测是否存在污染菌,采用宏基因组分箱方法能在种水平上精确评估益生菌种类,解析产品中的物种组成[11]。
流式细胞分析技术(flow cytometry analysis,FCM)是快速兴起的非培养活菌定量技术,通过荧光染料的选择可以快速区分活、死细胞,实现样品中活细胞的定量[12],但目前该检测技术很难实现每种菌的活菌计数。反转录荧光定量PCR(reverse transcription qPCR,RT-qPCR)可以实现活菌定量检测的目的[13],但RNA易降解,并且需将RNA反转录成cDNA后才能进行qPCR,操作过程复杂、容易带来误差,因此不适宜复杂样品中的微生物检测。叠氮溴化丙锭(propidium monoazide,PMA)是一种常见的活菌染料,其与qPCR结合的技术(propidium monoazide-quantitative PCR,PMA-qPCR),不仅可以特异性地定量菌株数量,而且可以区分活细胞和死细胞[14],作为靶向方法具有良好的应用潜力,已成为益生菌产品中乳酸菌活菌数量检测的有效方法[15],在肠道环境益生菌活菌数检测[16]的研究中亦有广泛应用,在其他研究领域,如水环境[17]、生物膜[18]中活微生物的检测以及流行病学研究中亦有文献报道[19]。
本研究以2种市售复合益生菌固体饮料及其添加的5种(6株)益生菌为研究对象,采用宏基因组测序分析方法、ANI分析方法及PMA-qPCR方法,在菌种水平精确鉴定复合益生菌产品的物种组成,并对活菌数进行快速定量,为复合益生菌产品的质量控制提供技术参考,对促进益生菌在食品行业更加安全和广泛的应用具有重要意义。
1 材料与方法
1.1 材料
1.1.1 产品选择
选择2种市售复合益生菌固体饮料产品:Life·space益倍适®益生菌固体饮料(产品P1)由动物双歧杆菌乳亚种(Bifidobacteriumanimalissubsp.lactis)Bi-07(简称菌株Bi-07)、动物双歧杆菌乳亚种(B.animalissubsp.lactis)HN019(简称菌株HN019)、鼠李糖乳杆菌(Lactobacillusrhamnosus)HN001(简称菌株HN001)、发酵乳杆菌(Lactobacillusfermentum)CECT5716(简称菌株CECT5716)和短双歧杆菌(Bifidobacteriumbreve)M-16V(简称菌株M-16V)组成,每袋(1 g)添加活菌75亿CFU;Nature’s Bay天然博士®益生菌固体饮料(产品P2)由菌株HN019、植物乳杆菌(Lactobacillusplantarum)299v(简称菌株299v)、菌株HN001和菌株CECT5716组成,每袋(1 g)添加活菌≥60亿CFU。
乳杆菌属(Lactobacillus)微生物的表型和基因型具有多样化特征,基于已公布的大量乳杆菌属菌种的全基因组数据分析,原乳杆菌属被重新划分为25个属,乳杆菌属菌种分类学地位已发生重要变迁[20]。2020年6月更新的QPS名单已采纳了新的分类学名称,亦认可原分类学名称的使用。考虑到新分类单元的使用存在过渡期,本研究仍沿用之前的乳杆菌属国际分类体系。
1.1.2 主要试剂、仪器及培养基
DNA提取试剂盒(QIAGEN Dneasy Blood & Tissue DNA),QIAGEN公司;荧光定量PCR试剂盒(TaKaRa TB GreenTMPremix Ex TaqTM(Tli RNaseH Plus))、pMD-19T TA克隆载体(TaKaRa pMD-19T vector),宝生物工程(大连)有限公司;PMA,Biotium公司;DNA纯化试剂盒(Cycle-pure kit),OMEGA公司;限制性内切酶EcoRI,NEB公司;MRS琼脂培养基、强化梭菌培养基,BD公司。
BD1236核酸蛋白分析仪,Biodrop公司;TRIO48普通PCR仪,Biometra公司;Multiskan FC酶标仪,Thermo Fisher Scientific 公司;PT-H18A LED光敏仪,Biotium公司;Bead Ruptor 12破碎仪,OMNI公司;7500型定量PCR仪,ABI公司;Miseq PE30016Sr RNA基因扩增子测序平台、Hiseq 3000宏基因组测序平台,Illumina公司。
1.1.3 菌株及培养方法
产品声称添加的菌株:动物双歧杆菌乳亚种(B.animalissubsp.lactis)Bi-07、动物双歧杆菌乳亚种(B.animalissubsp.lactis)HN019、植物乳杆菌(L.plantarum)299v、鼠李糖乳杆菌(L.rhamnosus)HN001、短双歧杆菌(B.breve)M-16V和发酵乳杆菌(L.fermentum)CECT5716分别分离自单一菌株制备而成的商业化纯菌剂,16S rRNA 基因序列和持家基因序列鉴定结果表明6株菌在种(或亚种)水平上的分类学地位与商业化纯菌剂声称一致。
5种菌的模式菌株:动物双歧杆菌乳亚种(B.animalissubsp.lactis)CICC 24210T,短双歧杆菌(B.breve)CICC 6079T,鼠李糖乳杆菌(L.rhamnosus)CICC 6135T,植物乳杆菌(L.plantarum)CICC 6240T,发酵乳杆菌(L.fermentum)CICC 24209T均来自中国工业微生物菌种保藏管理中心(CICC)。
双歧杆菌属菌株和乳杆菌属菌株分别采用强化梭菌和MRS固体培养基在37 ℃下厌氧、避光培养。
1.2 实验方法
1.2.1 基因组DNA的提取
用于宏基因组测序的固体饮料样品微生物总DNA的提取,按照QIAGEN Dneasy Blood & Tissue 提供的说明书进行,使用1%的琼脂糖凝胶电泳检测DNA的提取质量,使用Biodrop测定DNA浓度和纯度。
用于PMA-qPCR检测的固体饮料样品微生物总DNA及菌株纯培养物的DNA提取,参考SCARIOT等[21]的方法,采用机械破碎生理盐水菌悬液获取DNA粗提液的方式进行提取。所有DNA溶液置于-20 ℃ 保存待用。
1.2.2 16S rRNA 基因高通量测序及生物信息分析
1.2.2.1 PCR扩增及Illumina Miseq测序
使用338F (5′-ACTCCTACGGGAGGCAGCAG-3′)和806R(5′-GGACTACHVGGGTWTCTAAT-3′)引物对16S rRNA基因V3-V4可变区进行PCR扩增。将同一样本的PCR产物(3个平行样品)混合,以2%琼脂糖凝胶检测PCR产物,用胶回收试剂盒(axygen biosciences AxyPrep DNA gel extraction kit)进行PCR产物纯化,2%琼脂糖凝胶电泳检测回收效果。用QuantusTMFluorometer(美国Promega)对回收产物进行定量。使用NEXTFLEX Rapid DNA-Seq Kit进行建库,根据说明书进行操作。利用Illumina公司的Miseq PE300平台进行测序(上海美吉生物医药科技有限公司)。原始数据上传至NCBI SRA数据库(序列号:SRR15195391、SRR15195390)。
1.2.2.2 数据处理及分析
使用fastp[22](https://github.com/OpenGene/fastp,version 0.20.0)软件对原始测序序列进行质控,使用FLASH[23](http://www.cbcb.umd.edu/software/flash,version 1.2.7)软件进行拼接。使用UPARSE[24]软件(http://drive5.com/uparse/,version 7.1),根据97%[24]的相似度对序列进行操作分类单元(operational taxonomic units,OTU)聚类并剔除嵌合体。利用RDP classifier[25](http://rdp.cme.msu.edu/,version 2.2)对每条序列进行物种分类注释,比对Silva 16S rRNA基因数据库(v138),设置比对阈值为70%。
1.2.3 鸟枪法宏基因组文库构建及测序
使用NEXTFLEX Rapid DNA-Seq(美国,Bioo Scientific)建库,根据说明书进行文库制备。使用Illumina Hiseq 3000(美国Illumina)测序平台进行宏基因组测序(上海美吉生物医药科技有限公司)。原始数据已提交至NCBI SRA数据库(序列号:SRR15194982、SRR15194981)。
1.2.3.1 数据质控及拼接组装
使用fastp[22](https://github.com/OpenGene/fastp,version 0.20.0)对reads 3′端和5′端的adapter序列进行质量剪切,并去除剪切后长度小于50 bp、平均碱基质量值低于20以及含N碱基的reads,保留高质量的pair-end reads 和single-end reads。
1.2.3.2 基因组重建
采用MetaWRAP宏基因组分析流程对clean reads 进行分箱(Bining)操作。首先,利用read_qc模块对clean reads进行质控;其次,用assembly模块对质控后的宏基因组数据进行组装;再用binning模块对组装后的contigs进行分箱操作[26]。利用checkM对分箱后bins的完整度和污染度进行评估[27]。去除完整度<70%和污染度>5%的bin,对分箱结果再次提纯优化,利用blobology模块进行可视化分析[26]。
1.2.4 ANI分析
从NCBI genome数据库下载5个种的模式菌株的基因组序列,利用ANI Calculator软件分别对6个菌株与所在种内模式株的基因组序列进行ANI分析。以95%~96%的ANI值作为细菌物种界定的标准[9],对6个菌株进行物种鉴定。
1.2.5 PMA-qPCR 方法
1.2.5.1 PMA处理条件
参考GOBERT等[28]的方法制作热致死菌体,略有改动。刮取在37 ℃厌氧条件,固体培养48 h的乳酸菌,用无菌生理盐水稀释至108CFU/mL的菌悬液(OD620为0.4左右),分成2份,一份记为活菌,一份于80 ℃水浴处理20 min,取出立即于冰浴冷却,记为死菌(利用平板菌落计数法来检测无活菌生长)。
分别向500 μL活、死菌菌液中加入PMA溶液,使其终浓度为50 μmol/L,暗孵育5 min,每隔1 min混匀1次,于LED光敏仪曝光15 min。12 000 r/min室温离心15 min收集菌体,用于纯培养物DNA提取。通过qPCR检测PMA处理条件计算对死菌DNA扩增的抑制率,如公式(1)所示。

(1)
式中:C死菌-PMA(+),样品PMA处理的菌浓度,CFU/mL;C死菌-PMA(-),样品未经PMA处理的菌浓度,CFU/mL。
1.2.5.2 引物筛选及特异性分析
通过文献检索5种乳酸菌常用引物,先通过NCBI BLAST数据库(https://blast.ncbi.nlm.nih.gov/Blast.cgi)检索引物特异性,由生工生物工程(上海)股份有限公司进行引物合成,以1.1.3中的6株细菌为目标菌株,通过直接热裂解菌落的方式获取DNA,PCR 扩增验证引物特异性。进一步通过qPCR验证产物是否单一,引物来源及详细信息见表1。
1.2.5.3 qPCR标准曲线的建立
乳杆菌和双歧杆菌分别接种于MRS和强化梭菌固体培养基,于37 ℃厌氧培养24~48 h。分别刮取菌株HN001、菌株CECT5716、菌株299v、菌株Bi-07和菌株HN019的菌体,用无菌生理盐水调节OD620至0.4 左右,通过平板菌落计数法测定初始菌液浓度(CFU/mL)。取500 μL菌液,按照1.2.1的方法提取DNA;获得的基因组DNA以10倍梯度稀释至10-7,通过qPCR测定每个稀释度DNA对应的Cq值,以菌液浓度的对数值(lg CFU/mL)为x轴,Cq值为y轴,建立基于CFU的qPCR标准曲线。通过公式(2)计算qPCR扩增效率:
E=10-1/s-1
(2)
式中:s为标准曲线斜率。
刮取半环短双歧杆菌(B.breve)菌体,提取DNA,用qPCR引物进行PCR扩增获得目标产物,利用Cycle clean试剂盒纯化PCR产物。将纯化的片段连接到pMD-19T Vector,连接产物转化至感受态细胞Trans 5α,获得携带目的序列的阳性克隆。提取质粒DNA,用限制性内切酶EcoRI对质粒进行线性化,用Cycle clean试剂盒纯化酶切产物,测定线性化DNA浓度。通过公式(3)将DNA浓度换算基因拷贝数浓度。
(3)
式中:NA,阿伏伽德罗常数,6.02×1023。
对线性质粒进行梯度稀释,选择10-2~10-9作为模板进行qPCR,根据每个稀释度对应的质粒拷贝数的lg值(横坐标)和Cq值(纵坐标)绘制标准曲线,计算qPCR扩增效率。
1.2.5.4 qPCR 反应体系及反应条件
qPCR 反应体系:参考SYBR Premix ExTaqTM说明书提供的反应体系:SYBR qPCR Mix 12.5 μL、上、下游引物(10 μmol/L)各1.0 μL、50×Rox 0.5 μL、稀释的DNA模板5 μL,加无菌ddH2O补足体积至25 μL。
qPCR反应条件:每对引物的反应条件略有不同,详细信息见表1。循环结束后进入溶解曲线分析,程序为65 ℃ 逐步升温至95 ℃,每升温0.5 ℃检测一次荧光信号,每次检测持续5 s。
1.2.6 PMA-qPCR 定量检测产品中的活菌数
根据产品声称的活菌总数,加入适当体积的生理盐水,使活菌浓度为108CFU/mL,混匀并充分溶解菌粉;取500 μL菌液于1.5 mL无菌离心管中,12 000 r/min室温离心15 min收集菌体,去掉上清液;再用500 μL的生理盐水清洗菌体1次;用500 μL生理盐水充分悬匀菌体,按照1.2.5进行PMA处理;按照1.2.1的方法提取基因组DNA,通过qPCR定量检测产品中每种菌的活菌数。
1.2.7 平板菌落计数
按照1.2.6的方法溶解菌粉;对菌液进行梯度稀释,选取2~3个连续的适宜稀释度菌液1 mL于灭菌平皿内,每个稀释度做2个平行实验,于37 ℃ 厌氧、避光培养72 h后进行菌落计数。
2 结果与分析
2.1 产品中微生物多样性分析
通过基于16S rRNA 基因的338~806 nt区域进行PE300高通量测序,分析产品P1和P2中的微生物组成。过滤后,P1和P2的序列数分别为61 338和69 853条(表2)。序列平均长度为424 bp,每种产品的有效序列均超过50 000条,且稀释曲线均趋于平稳,表明测序数据质量满足微生物多样性分析的要求。
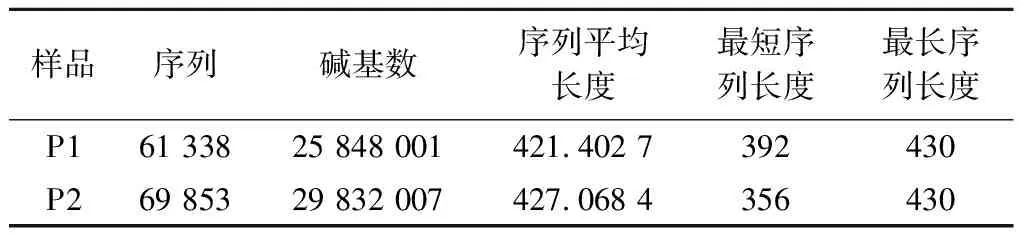
表2 产品16S rRNA基因扩增子测序 信息统计 单位:bp
每个产品的测序结果分析均只产生4个OTU,表明产品P1和P2中均存在4种菌,与P1和P2声称添加的菌种组成一致。对获得的OTU进行注释,在种水平上,除一种双歧杆菌注释至属(unclassifiedBifidobacterium)水平外,P1中含有的其他3种菌分别注释为发酵乳杆菌(L.fermentum)、鼠李糖乳杆菌(L.rhamnosus)、短双歧杆菌(B.breve);P2中含有的其他3种菌分别注释为发酵乳杆菌(L.fermentum)、鼠李糖乳杆菌(L.rhamnosus)、植物乳杆菌(L.plantarum),均与产品声称的菌种一致。根据产品说明显示的物种添加信息,推测是由于动物双歧杆菌乳亚种的种间相似性高,短片段的16S rRNA 基因无法将其界定到种水平。为了在种水平上精确鉴定产品中的每个物种,同时对产品DNA进行宏基因组测序。
2.2 产品中的物种鉴定
2.2.1 益生菌基因组重建
对基于Illumina Hiseq 3000测序平台的宏基因组数据,采用fastp软件、碱基质量值≥20,对Raw reads进行质控,质控后产品P1和P2的clean reads 数分别为98 414 620和99 779 276条,每种产品质控后的clean reads均占Raw reads的98.77%以上(表3)。
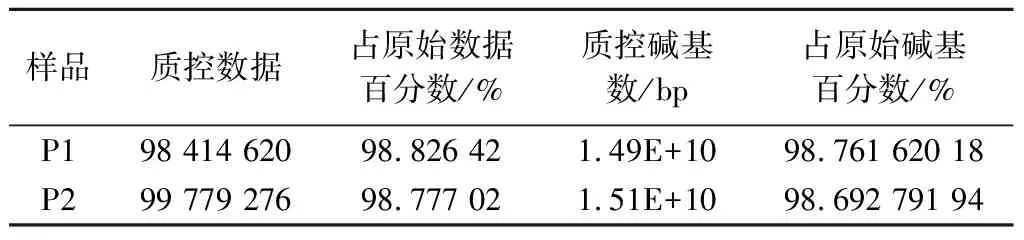
表3 产品宏基因组测序质控后数据统计Table 3 Data statistics after metagenomic sequencing and quality-control
采用MetaWRAP宏基因组分析流程,用maxbin2算法对组装后的contigs进行分箱(binning),每个产品的contigs数据均被分为4个bins(表4),与OTU数和产品添加的菌种数一致。
利用checkM对分箱后bins的完整度和污染度进行评估,结果表明,P1和P2分箱产生的bins的完整度均高于98%,污染度均低于5%(表4),成功实现了P1和P2宏基因组数据的分箱。每个bin分别通过blast与NCBI数据库进行比对,初步分析每个bin所代表的物种(图1),与16S rRNA基因微生物多样性一致。宏基因数据分箱成功实现4种菌的基因组重建。
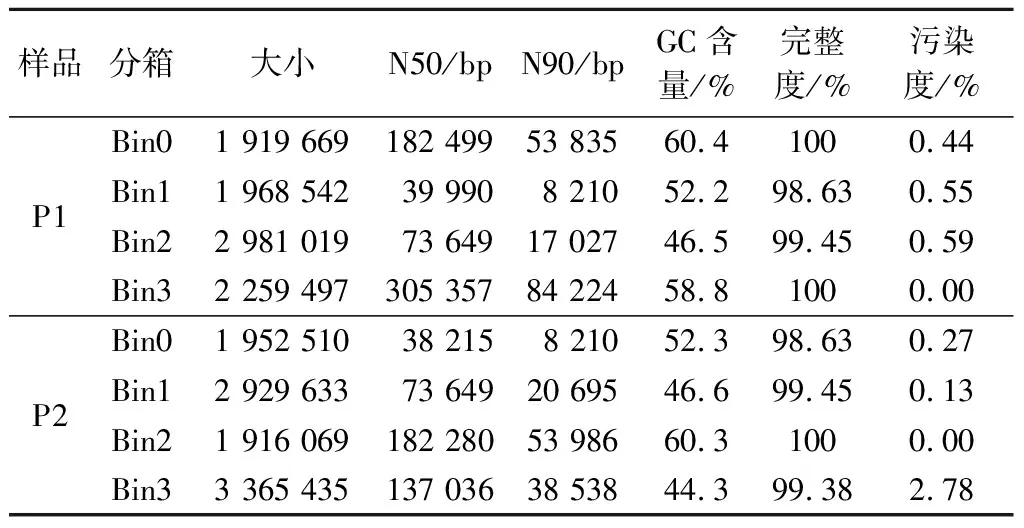
表4 宏基因组分箱结果Table 4 The results of metagenomics binning
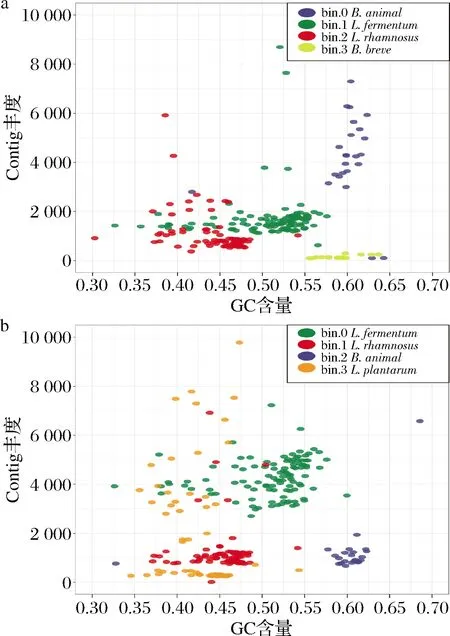
a-宏基因组分箱分析产品P1 的bins丰度; b-宏基因组分箱分析产品P2 的bins丰度图1 Bin丰度散点图Fig.1 Scatter plots of bins abundance
2.2.2 益生菌种水平鉴定
对分箱获得的bins与NCBI NT数据库进行比对,确定其代表的菌种。进一步通过bins与所代表种内的模式菌株的基因组进行ANI分析,每个样品中的4个bins与模式菌株基因组ANI分析的值均大于96%,表明每个bin代表的菌种与模式菌株为同一个种(表5)。产品P1中的bin 0与B.animalis种内的B.animalissubsp.lactisDSM 10140T相似性较高,ANI值为99.99%;产品P2中的bin 2与B.animalis种内的B.animalissubsp.lactisDSM 10140T相似性较高,ANI值亦接近99.99%;表明P1中的bin 0和P2中的bin 2代表的均为B.animalissubsp.lactis(表5),与产品声称一致。通过circos v 0.69-8 软件对产品中的bins进行可视化,每个bin中挑选最长的12条contigs进行作图(图2)。

表5 Bins与参考模式菌株基因组ANI 分析结果Table 5 The analysis results of ANI between bins and type strains genomes
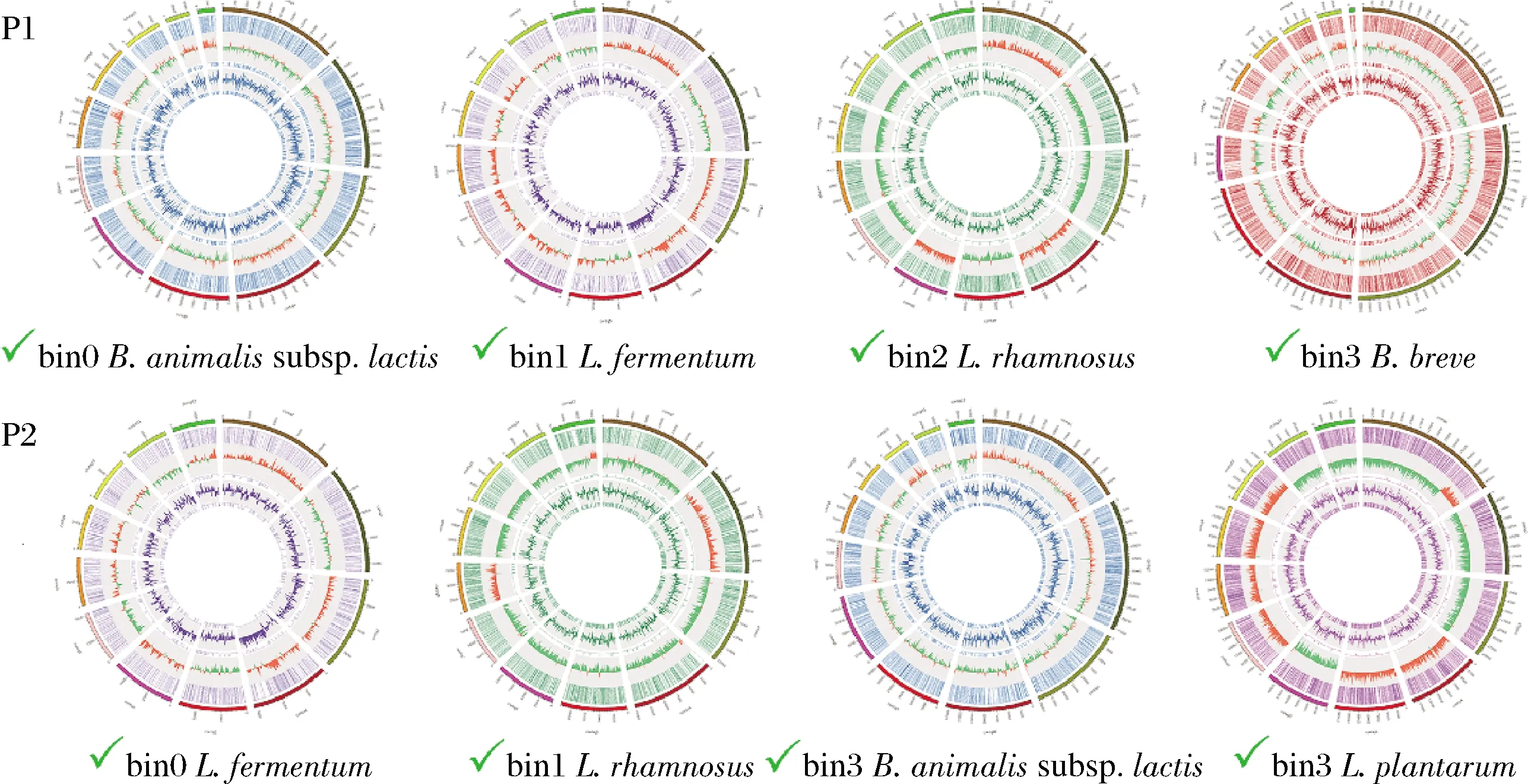
图2 通过宏基因组分箱进行产品菌种基因组重建Fig.2 Re-constructed genomes from probiotic products by means of the metagenomics binning 注:每一个产品的bins 都由横向排列的遗传图谱来表示;相同物种用同一种颜色显示,绿色“√”表示分箱得到的物种与产品声称菌种一致
2.3 建立PMA-qPCR 检测体系
2.3.1 引物特异性分析
引物的特异性结合是qPCR检测生物量准确度的关键。通过NCBI BLAST数据库对文献中筛选的引物特异性进行初步判断,进而通过普通PCR扩增检测引物的特异性,最后通过qPCR溶解曲线判断产物的单一性。如表6所示,发酵乳杆菌引物(LFer-1/LFer-2)、植物乳杆菌引物(LplantarumF/LplantarumR)、鼠李糖乳杆菌引物(LrhamnosusF/LrhamnosusR)在种间、自身基因组中均具有高特异性,每株菌基因组DNA的PCR扩增条带的大小与模式菌株(阳性对照)基因组DNA、靶菌株基因组DNA及对应产品总基因组DNA的PCR扩增产物条带大小一致,并且qPCR溶解曲线为单一峰;短双歧杆菌引物(BiBRE-1/BiBRE-2)以菌株HN019基因组DNA为模板时,会产生弱的条带,动物双歧杆菌乳亚种引物(Bal-talF/Bal-talR)在以菌株299v基因组DNA为模板时,亦会产生弱的条带,但两条带的大小与模式菌株(阳性对照)基因组DNA、靶菌株基因组DNA及对应产品总基因组DNA的PCR扩增条带大小不一致,并且产品qPCR溶解曲线单一,不影响P1中短双歧杆菌和P2中动物双歧杆菌乳亚种的准确定量。
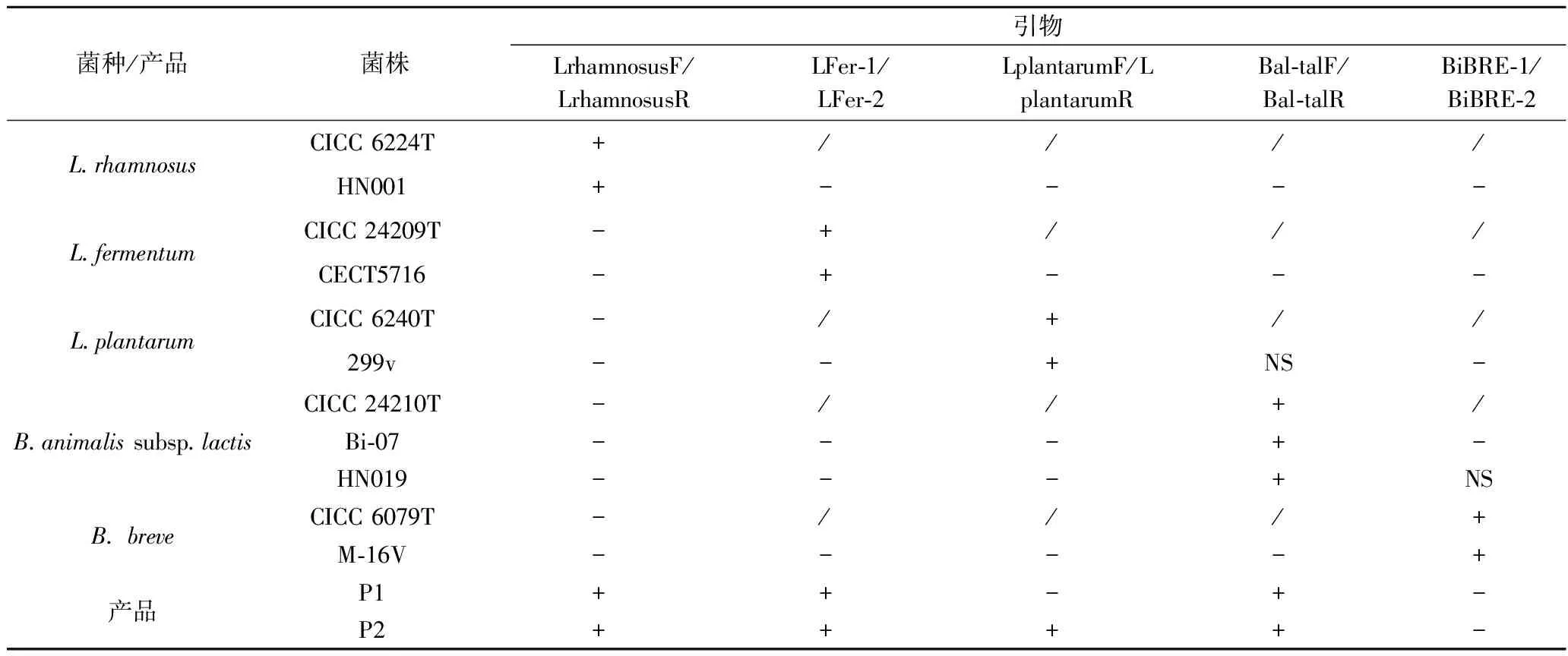
表6 qPCR 引物特异性检测Table 6 The specificity test of qPCR primers
2.3.2 建立定量PCR 标准曲线
以2种产品中的6株菌为研究对象,分别建立每株菌的qPCR标准曲线。如表7所示,qPCR反应信号Cq值与菌落浓度的对数值(lg CFU/mL)之间线性关系良好(R2>0.98);通过优化反应体系与条件,扩增效率均达到85%~110%,标准曲线的检测限为102CFU/mL(表7)。
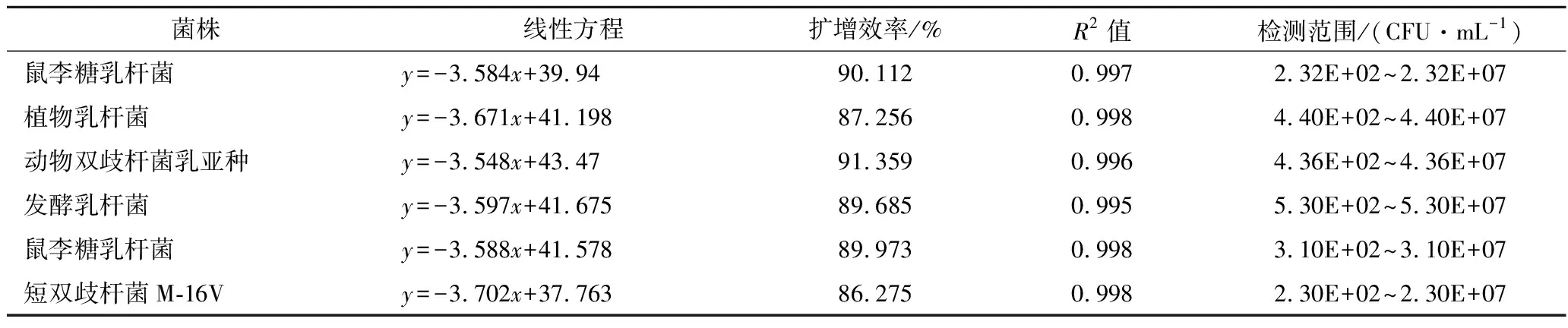
表7 qPCR标准曲线参数信息Table 7 The parameter information of standard curves for qPCR
2.3.3 PMA条件确定
因不同微生物对PMA的敏感度不同[33],采用qPCR方法检测PMA作用条件对6株乳酸菌死菌DNAqPCR扩增的抑制,以及对活菌DNA qPCR的影响。参考益生菌及致病菌活菌数定量研究中最常用的PMA作用条件[30]:PMA终浓度为50 μmol/L,暗孵育时间5 min,曝光时间15 min,作为最初的PMA作用条件。
2.3.3.1 PMA处理对活菌qPCR扩增影响
取新鲜的纯培养物,调整菌液浓度至108CFU/mL左右,通过qPCR检测PMA作用条件对活菌DNA qPCR的影响。通过t检验分析不加PMA处理的活菌与加PMA处理的活菌qPCR的Cq值,结果表明,6株菌的活菌在有和无PMA处理后,其qPCR 的Cq 值间均无显著性差异(P>0.05)(图3-a),表明在有、无PMA处理的样品中可扩增的靶DNA量基本相等,表明此PMA作用条件不影响活菌qPCR扩增。
2.3.3.2 PMA处理对死菌qPCR 抑制
通过1.2.5.1中的公式计算PMA作用条件对死菌DNA 的qPCR 扩增抑制率。结果表明,6株菌的抑制率均高达99.11% 以上(图3-b)。最终通过qPCR确定,除菌株CECT 5716 PMA作用终浓度为40 μmol/L外,其他5个菌株的PMA作用终浓度均为50 μmol/L;所有菌株PMA处理的暗孵育时间均为5 min,曝光时间均为15 min。
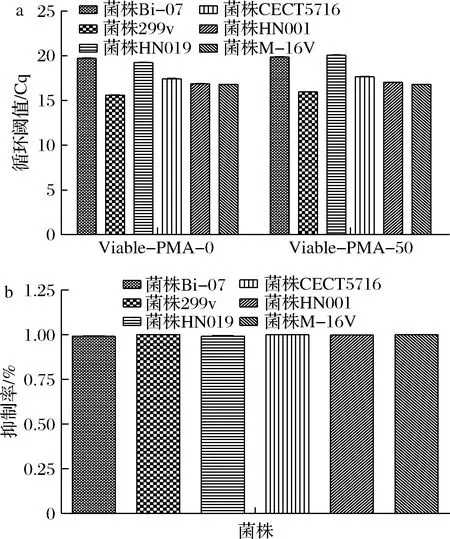
a-50 μmol/L PMA对活菌DNA的PCR扩增影响; b-50 μmol/L PMA对死菌DNA的PCR扩增抑制率图3 PMA作用条件对活菌和死菌DNA qPCR的影响Fig.3 Effect of PMA treatment on DNA qPCR of viable and non-viable bacteria
2.3.4 PMA-qPCR检测方法的可靠性评估
调整纯菌株培养物的菌液浓度至108CFU/mL,基于建立的qPCR标准曲线,计算每株菌的活菌数,并与菌落计数结果进行比较。如表8所示,t检验分析2种活菌计数方法,结果无显著性差异(P>0.05)。表明在菌液浓度为108CFU/mL左右时,PMA-qPCR方法具有良好的可靠性。
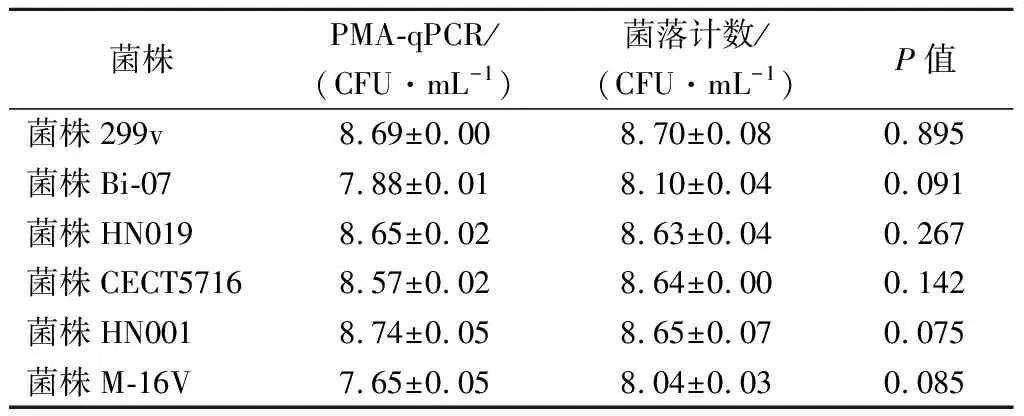
表8 PMA-qPCR与平板菌落计数法对6株 菌活菌数的检测结果Table 8 Detection results of six strains of viable bacteria by PMA-qPCR and colony counting
2.4 PMA-qPCR检测方法在产品中的应用
复合益生菌产品是一种由多菌种混合的食品,检测其活菌数是判断产品质量的关键。在种水平检测复合益生菌产品中每种菌的含量仍具有一定难度。目前,关于PMA-qPCR方法用于益生菌及致病菌靶菌种的活菌定量检测的研究较多。本研究亦采用PMA-qPCR检测法,在种水平上对市售2种固体饮料P1和P2中的6株益生菌的活菌数进行定量检测。
如图4所示,将产品P1中每种菌的PMA-qPCR定量结果相加获得产品P1的活菌总数为1.01×1010CFU/g,t检验分析其与平板菌落计数结果1.01×1010CFU/g无显著性差异(P=0.78>0.05)。产品中B.animalissubsp.lactis和L.rhamnosus含量较高,分别为4.86×109和4.33×109CFU/g;其次为L.fermentum3.95×108CFU/g,3种益生菌活菌数与产品实际添加数量基本吻合。B.breve的含量最低,约为6.38×106CFU/g,在纯菌株培养过程中发现短双歧杆菌M-16V对环境敏感,易死亡,PMA-qPCR检测结果亦显示短双歧杆菌活菌数量低于实际添加量,推测短双歧杆菌M-16V的不稳定性是导致其活菌检出量低的主要原因。
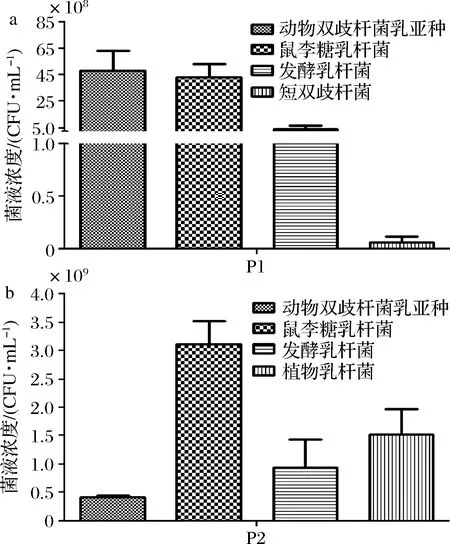
a-产品P1;b-产品P2图4 PMA-qPCR 法检测产品中6 株乳酸菌的含量Fig.4 Quantitative test results of six strains of lactic acid bacteria in products by PMA-qPCR
将产品P2中每种菌的PMA-qPCR 定量结果相加获得产品P2的活菌总数为6.00×109CFU/g,t检验分析其与平板菌落计数结果4.35×109CFU/g无显著性差异(P=0.427>0.05)。产品中L.rhamnosus含量最高,为3.12×109CFU/g;其次为L.plantarum和L.fermentum,活菌数分别为1.53×109和9.36×108CFU/g;B.animalissubsp.lactis含量最低,约为4.16×108CFU/g,4种益生菌活菌数量与产品实际添加数量基本吻合。
3 结论
复合益生菌产品的菌种组成与活菌数的含量是产品质量的关键,本研究以2种复合益生菌固体饮料及其添加的5种(6株)益生菌为研究对象,采用高通量测序及组学分析技术实现了2种复合益生菌产品物种组成的精确鉴定;通过PMA-qPCR方法实现了产品中每种乳酸菌活菌数快速定量检测。采用多技术联用,构建适合益生菌产业应用的快速、精准的活菌计数及检测技术体系已成为益生菌行业的重要研究方向,本研究为建立和完善复合益生菌产品的质量控制提供技术支撑,有助于提升产品评价、质量控制及市场监管水平。
猜你喜欢
当代水产(2022年3期)2022-04-26
中国典型病例大全(2022年7期)2022-04-22
中国乳品工业(2022年3期)2022-04-11
河南畜牧兽医(2021年9期)2021-12-10
当代水产(2021年2期)2021-03-29
保鲜与加工(2021年1期)2021-02-06
乳业科学与技术(2020年4期)2020-12-10
中华养生保健(2020年2期)2020-11-16
消费者报道(2019年3期)2019-06-12
消费导刊(2018年8期)2018-05-25
