
微量元素铜与人体生理功能和疾病
2022-05-12 07:52向思佳刘扬中
大学化学 2022年3期
向思佳,刘扬中
中国科学技术大学化学与材料科学学院,合肥 230026
铜是生物体必不可少的微量元素,成人体内含有100-200 mg铜,是仅次于铁和锌的第三微量元素。铜的价态通常是+1价和+2价,在细胞中铜不会以自由离子形式存在。细胞内的铜处于一个相对稳定的状态,其中铜转运蛋白(hCtr1)的铜摄取和ATP酶(ATPase)的铜外排对维持胞内铜的正常水平起到至关重要的作用。当铜缺少或过量时,就会导致疾病的产生甚至是威胁生命;另外,利用关键蛋白对铜的依赖性,可以通过调控体内铜的水平进行癌症等疾病的治疗。本文从铜的配位结构、相关蛋白功能对相关问题进行阐述。
1 铜的配位化学
Cu的价层电子结构为3d94s2,所以通常呈现出的价态为+1价(3d10)和+2价(3d9)。不同价态的铜可以形成不同的配位结构,一价铜通常形成线性、平面三角形或四面体的结构;而二价铜则通常形成平面四边形、四角锥或八面体。根据软硬酸碱的概念,Cu(I)属于软酸,而Cu(II)属于中间态酸。根据软硬酸碱理论,软酸与软碱或硬酸与硬碱具有较强的亲和性。因此,Cu(I)更倾向于与半胱氨酸、蛋氨酸配位,而Cu(II)则倾向于与组氨酸配位。这些配位性质使得铜在蛋白质中有着三种特征配位方式:
(1)以四面体作为配位结构的I型铜(图1a):最常见的是His2CysMet,其具有相对高的还原电势,600 nm附近强吸收,通常与电子传递相关。质体蓝素便是属于这一类铜蛋白。
(2)以平面四边形为配位结构的II型铜(图1b):通常与N或N/O配位。铜锌超氧化物歧化酶,多巴胺氧化酶都是属于这一类的铜蛋白。
(3)以双核铜为配位结构的III型铜(图1c):配位原子为(His)3Cu-X-Cu(His)3,两个Cu通过桥联原子共价偶合重叠,常见于氧化酶或O2运输蛋白。血蓝蛋白、酪氨酸酶便是属于这一类铜蛋白。
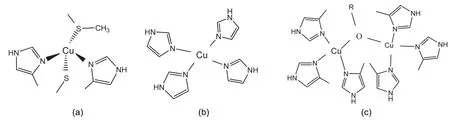
图1 铜蛋白中三种典型的Cu(II)配位结构
2 人体中重要的铜蛋白
人体中部分蛋白质依赖于铜的配位而发挥其正常功能,这类蛋白就是铜蛋白。下面介绍几种重要的铜蛋白。
2.1 血浆铜蓝蛋白(CP)
血浆铜蓝蛋白由肝脏细胞制造,核心位置包含六个铜离子,在星形胶质细胞中表达的CP是以GPI锚定形式存在的,主要作用是进行铁和铜的氧化还原反应,反应时二价铜离子还原成一价,而把铁离子由二价氧化成三价,并协助转铁蛋白运输铁[1],属于亚铁氧化酶。
CP-转铁蛋白系统是细胞内铁的主要输出途径,血浆中的CP可以利用其亚铁氧化酶活性将Fe2+氧化成能与转铁蛋白结合的Fe3+,Fe3+结合转铁蛋白后被转运至靶细胞,经内吞后进行代谢或者储存。结合了铜的CP是血清中铜的主要载体,其携带了血浆中95%的铜,所以可以作为铜含量的指示物,即通过测定CP含量来判断机体中铜的含量,进而诊断某些肝、胆、肾疾病。当铜蓝蛋白到达靶细胞表面时,会与其相应的受体相互作用释放铜,这些铜会被靶细胞吸收和利用,通过CP对铜离子的结合和释放,可以实现铜在不同的组织器官中有不同的分布。
当铜蓝蛋白缺乏,或由于基因突变导致其亚铁氧化酶活性丧失,如遗传性铜蓝蛋白缺乏症,会使Fe2+无法氧化成Fe3+,进而不能与铁转运蛋白结合而进行铁的输出,引发缺铁性贫血,同时也会导致神经系统中Fe2+大量沉积和Fe3+缺乏,从而引起神经系统退行性疾病[2]。
2.2 细胞色素C氧化酶(CCO)
细胞色素C氧化酶作为二聚体线粒体膜蛋白靠近线粒体内膜,是一种电子转移蛋白,在生物体的氧化呼吸链中,能将电子传递给氧气生成水,且能实现质子的跨膜转移[3]。作为一种跨膜蛋白,通过α螺旋贯穿线粒体内膜,而在膜内或膜外则是充满α螺旋和β折叠结构。细胞色素C氧化酶的金属离子中心由3Cu、2Fe构成,两个Fe通过卟啉环配位构成了细胞色素a和细胞色素a3,两者结合紧密,不可分开;而三个Cu则是构成了一个双核CuA中心和一个单核CuB中心。
细胞色素C氧化酶在电子转移过程中会发生反应:

电子转移途径为:
CytC → CuA→ Cytochrome a → Cytochrome a3-CuB→ O2
铜是细胞色素C氧化酶中重要的金属中心,当铜缺乏时,细胞色素C氧化酶的活性会被抑制,从而使氧化呼吸链无法正常进行,机体无法正常产生能量。
2.3 铜锌超氧化物歧化酶(CuZn-SOD)


催化机理为:
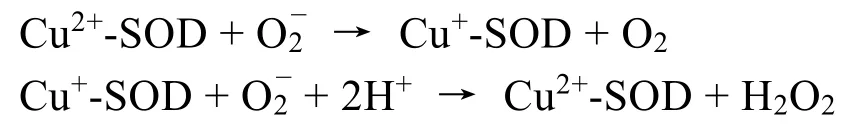
SOD的金属中心含有Zn和Cu两种金属离子,其中Zn主要是起到结构稳定的作用,而Cu则主要起到催化作用,这一点从机理上也可以看出。由于人体内在或外界的因素,会造成机体、皮肤组织的自由基导致皮肤的衰老,SOD可以消除氧自由基,从而抑制或缓解机体的衰老;此外,氧自由基会导致脂质过氧化物积累,引起动脉粥样硬化等疾病,因此SOD对高血脂引起的心脑血管疾病有预防作用;同时,SOD对于各类自身免疫性疾病都有一定的疗效,如红斑狼疮、硬皮病、皮肌炎等[4]。
3 铜的摄取、调控和代谢
3.1 铜的摄取
铜首先由肠上皮细胞(IECs)摄取,进入细胞后结合于Cu相关蛋白质,通过肠侧膜进入外周循环。随后铜被输送到肝脏,在这个维持铜稳态的中枢器官中,铜被肝细胞整合到分泌的血浆铜蓝蛋白(CP)中,该酶可催化血清中亚铁氧化成三价铁,使其能够与转铁蛋白结合,从而实现铁离子的细胞摄取[5]。下面具体介绍哺乳动物中铜离子的细胞摄取过程。
铜转运蛋白(Ctr1)承担起了跨膜运输铜的重任:人类铜转运蛋白(简称hCtr1)是由190个氨基酸残基构成的三跨膜蛋白,N-端在膜的外侧,C-端在膜的内侧,N-端有组氨酸富集和蛋氨酸富集的基序各两个[6]。通常hCtr1是由三聚体的模式聚合,在高分辨率的电镜下可以观测到对称的通道状三聚体,形成了一个“离子选择过滤器”,选择性摄取Cu+离子。Ctr1膜外N-端的金属结合域捕获Cu+离子,Cu(I)通过Cu-S键配位结合于hCtr1,通过通道内的金属配位氨基酸进入细胞[7]。通过突变或截短hCtr1序列发现,N-端、C-端的金属结合域均对铜的摄取有影响[8]。
有趣的是,除了Cu可以利用Ctr1被细胞摄取,一些金属药物也可以利用Ctr1,如顺铂就是利用了铜转运蛋白hCtr1进入细胞中从而起到诱导肿瘤细胞凋亡的作用。
3.2 铜的胞内组装
铜进入细胞后与铜伴侣蛋白结合,并被组装到功能蛋白中,以防止游离的铜离子催化胞内生物分子氧化,产生活性氧(ROS)。如SOD发挥酶的功能需要铜离子,但细胞内并不存在自由的铜离子,所以需要利用伴侣蛋白提供铜离子,促使SOD成熟。伴侣蛋白为细胞内金属离子运输、分配、平衡起到重要的作用,这一转运体系受损可能导致疾病的发生。下面介绍三种经典的铜伴侣蛋白。
3.2.1 CCS:铜锌-超氧化物歧化酶的铜伴侣蛋白
CCS蛋白(Copper Chaperone for Superoxide Dismutase),是第一个被发现的铜伴侣蛋白,在哺乳动物细胞和酵母细胞内为SOD蛋白提供铜离子。酵母细胞实验表明,敲除yCCS不影响细胞中Cu和SOD蛋白的含量,但会降低细胞内SOD蛋白的活性;而通过体外实验发现自由的Cu2+离子(Cu-GSH)可以激活SOD,铜螯合剂BCS(bathocuproine sulfonate)能够抑制Cu-GSH的作用;但Cu-CCS可以激活SOD且不受BCS影响。根据配位结合常数计算表明,细胞的自由铜离子浓度不会超过10-18mol·L-1,如果一个细胞内含有一个铜离子,则浓度为10-10mol·L-1,因此细胞内不存在自由的铜离子。同时这一结果也说明了CCS对激活SOD的必要性[9]。
3.2.2 Atox1:ATPase的铜伴侣蛋白
Atox1由68个氨基酸残基组成,有两个保守序列:一个是金属结合位点,另一个是核定位信号。Cu的配位可以通过Cys-Cu(I)使蛋白二聚:如果二聚的蛋白是两个Atox1,通过其上的核定位信号介导蛋白质由细胞质向细胞核内转运,在细胞核内,Atox1可以作为cyclin D1的转录因子,激活CCND1的启动子,进而促进CCND1的表达,其可以通过激活CDK激酶蛋白,在细胞周期中起调控作用[10];如果二聚的蛋白是一个Atox1和一个ATPase,Cu(I)可以通过改变与其结合的Cys-Cu(I)共价键,从而将Cu(I)从Atox1传递给ATPase。ATPase就是ATP酶,可以催化水解ATP成ADP和Pi,也可以作为一种铜转运体,将铜运出或运进细胞。
铜转运型ATPase分为ATP7A和ATP7B两种。ATP7A存在于肝脏之外的多数细胞中,ATP7A在小肠中帮助细胞摄取铜。其他细胞中的ATP7A具有双重功能,通常存在于高尔基体中的ATP7A为酶、骨、发、神经系统等提供铜;当细胞内铜过多时,ATP7A蛋白会离开高尔基体,转移至细胞膜,将铜泵出细胞。门克斯病(Menkes disease)是一种罕见的基因疾病,由于ATP7A突变导致小肠的铜吸收受阻,患者体内多种生理功能无法进行[11]。而ATP7B主要存在于肝脏,少量在肾、大脑中。在肝脏中,ATP7B通常存在于高尔基体,为血浆铜蓝蛋白提供铜,再通过血液转运至其他组织;当肝脏铜过多时,ATP7B蛋白离开高尔基体,将铜泵出至囊泡,通过胆汁排出。如果ATP7B基因突变,会导致肝豆状核变性疾病(又称威尔逊病,Wilson disease),细胞内铜分配不正常,使得铜过量[12]。因此,Atox1作为一种伴侣蛋白,对于铜在细胞中的整体分布起到至关重要的作用。
3.2.3 Cox17:细胞色素C氧化酶的铜伴侣蛋白
在真菌中,首次发现了Cox17是细胞色素氧化酶功能所必须的,是一种富含半胱氨酸的蛋白质,之后研究发现,在哺乳动物中同样需要Cox17,这是激活细胞色素C氧化酶(CCO)、促进胚胎发育所必需的蛋白质[13]。Cox17中6个Cys都是铜的结合位点,完全氧化的时候形成3个二硫键,不能结合金属,部分氧化的Cox17有2个二硫键,可以结合1个Cu+离子,而还原态的Cox17没有二硫键,可以与4个Cu+离子协同结合,形成多核Cu(I)簇[14]。
Cu通过Cox17运输到线粒体,在额外的蛋白质(如SCOs和Cox17)的帮助下,Cu并入细胞色素C氧化酶。我们在研究中发现,Cox17还可以将抗肿瘤药物顺铂传递至线粒体,这一过程对顺铂促进细胞凋亡的药效具有重要的贡献。有趣的是,Cox17对顺铂的结合具有选择性。
3.3 铜的调控
由于铜的不可控积累可能会导致氧化应激或者与大分子结合不恰当,所以需要对铜的摄取、分布和使用进行严格的稳态调控。这里主要介绍铜摄取调控。
前文提到人体铜的摄取是通过铜转运蛋白hCtr1实现的,所以可以通过调节hCtr1的表达来控制铜的摄取。而hCtr1的表达则是由转录因子Sp1调控的:当胞内Cu不足时,Sp1会与hCtr1的启动子结合,增强hCtr1的表达,同时也能与Sp1的启动子结合,增强Sp1的表达,作为一种正反馈产生更多的hCtr1,促进Cu的摄取;当胞内Cu过量的时候,过量的Cu会取代Sp1锌指结构中的锌,改变Sp1的结构,破坏了Sp1对hCtr1启动子的识别,从而停止了Sp1和hCtr1的转录表达,从而使细胞对铜的摄取减弱,使细胞内铜水平降低[15]。而人体铜的排出则是通过ATPase实现的,当铜过量时,ATPase可以将Cu泵出细胞外,从而对细胞内铜的含量起到调控的作用。
4 铜与疾病
4.1 铜缺乏症
Menkes disease是一种罕见的伴X隐性遗传性疾病,病患儿出生时正常但多在2-4个月后就会开始出现严重的神经系统退化表现,平均生存年龄仅1.8岁,经组氨酸铜治疗可明显改善[11]。ATP7A的活性缺失导致铜不能从肠上皮细胞进入血液,因此以铜缺乏为特征。
铜缺乏的第一个临床表现是贫血;铜缺乏的下一阶段可能涉及结缔组织缺陷,甚至减少骨密度,而在铜缺乏症晚期,除了全血细胞减少外,还会有严重的神经病变[16]。由于铜向大脑的传递减少,Menkes disease患者会表现出严重的精神和发育障碍,部分原因是由于铜依赖性酶的活性降低,除了严重的进行性神经变性外,Menkes disease的临床特征还包括结缔组织异常,肌张力减退和皮肤和头发色素沉着减退。
线粒体功能受损是Menkes disease导致严重大脑损伤的主要因素。在Menkes disease病患者的脑中观察到细胞色素C氧化酶活性缺乏,大脑乳酸水平升高以及n-乙酰天冬氨酸含量降低,它们的合成与神经元线粒体能量的产生相关联,如此的变化可以作为线粒体氧化磷酸化失败的进一步指标。
注射给药给予铜化合物用于治疗Menkes disease可以改善临床结果,但至少要在新生儿出生后不久就开始治疗。而具有剩余ATP7A活性的Menkes disease新生儿比完全丧失功能的新生儿更容易对这种治疗做出反应,因此,恢复至少低水平功能的ATP7A的基因治疗可能成为一种代替的治疗方案[17]。
4.2 铜过量症
Wilson disease是一种以青少年为主的常染色体隐性遗传病,通常伴随与肝脏、眼、神经相关的症状,起源于铜运输ATP酶——ATP7B的遗传缺陷。由于ATP7B功能受损,铜不能从肝细胞分泌到胆汁并在肝脏和其他组织中积累,导致恶性、呕吐和腹泻等急性肠胃道症状,并产生慢性铜毒性。大约一半的Wilson disease患者会出现中枢神经系统毒性,而神经节和大脑协调运动的其他部分也可能会受到影响,产生运动障碍[16]。研究发现,患者通常会产生异常的星形胶质细胞即阿尔兹海默病I型和II型细胞,这可以作为Wilson disease典型的神经病理特征。
Wilson disease的治疗通常采用D-青霉胺或三乙基四曲胺等铜螯合剂进行治疗,以恢复和维持体内的铜稳态[17]。青霉胺是一种还原性螯合剂,其还原步骤可以降低铜对蛋白质的亲和力,使其更有效地被螯合,但青霉胺也有严重的副作用,会导致血液学或肾脏毒性。而对于未出现症状的患者,可以通过锌治疗,锌可以诱导肠道金属硫蛋白(MT)的合成而使机体缺铜,MT蛋白对于铜有很高的亲和力,并形成一种铜-蛋白复合物,这种铜复合物不能被吸收,并通过粪便排出,由于锌的毒副作用相对较低,它已经成为了Wilson disease病维持治疗的首选药物[16]。
4.3 铜与肿瘤
由于铜是许多必需酶的关键成分,为了满足肿瘤细胞快速增殖对铜的需求,癌症组织和癌症患者的血清铜水平升高。癌症的进展包括不受控制的细胞生长,随后是癌细胞的侵袭、传播和继发性肿瘤的形成。如果肿瘤生长得大于几毫米,就需要血管生成。铜对肿瘤的影响可以通过影响几种诱导促血管生成反应的分子途径,包括与血管生成因子的直接结合并促进他们的表达;也可以通过激活代谢来影响癌细胞的转移能力[18]。
研究发现,在恶性组织中,平均铜浓度比正常组织高出46%,尤其是在大肠、胃、膀胱和女性生殖器官中尤为明显。自由基,特别是超氧化物自由基离子被认为是癌症的可能原因。Cu水平显著增强会促进铜在线粒体中介导的催化作用,从而导致活性氧(ROS)的生成,过量的ROS会导致蛋白质的氨基酸侧链被氧化或肽链断裂从而改变蛋白质性质,导致各种酶活性的丧失;ROS可能氧化DNA的碱基或降解DNA;此外,ROS还可能作用于细胞膜的不饱和脂肪酸,造成脂质过氧化反应,破坏膜的正常功能[19]。这些过程都可能引发细胞生长状态的改变,所以铜和肿瘤关系十分密切[20]。
铜在促进生理血管生成和恶性血管生成方面也发挥重要的作用。研究表明:实体肿瘤的生长和转移能力依赖于血管的生成,而血管内皮生长因子(VEGF)是血管生成的关键因子,且铜被发现参与了这样一些在肿瘤血管生成中的因子的活性调节。由于肿瘤可以通过形成新血管使自身生长、侵袭和转移,所以一种抑制肿瘤的抗癌方法就是降低铜的含量,一个很经典的药物就是铜螯合剂四硫钼酸盐(TM),其可以快速地螯合消耗铜,是一种有效的抗血管生成剂。TM有双重的作用机制:在食物中加入可以使TM、铜和食物中的蛋白形成三方复合物,从而防止铜的吸收;在两餐之间服用,TM被吸收到血液中,并与白蛋白以及血清铜形成三方复合物,这种络合铜无法被细胞吸收,从而使得铜量迅速减少[16]。
铜螯合疗法的优点之一是,它减少了许多因子的分泌,潜在地解决了血管生成的冗余问题,在炎症性乳腺癌细胞系中,用铜螯合剂治疗后,机体释放的血管生长因子如VEGF、FGF、IL-6等水平显著降低,与未治疗的乳腺癌细胞培养基相比,血管生长显著减少[21]。
最近的研究发现,Atox1具有与癌症进展相关的转录活性。上文提到Atox1可以作为一种铜依赖转录因子促进细胞周期蛋白D1的表达,这是一种参与细胞周期和细胞增殖的关键蛋白[18]。最近发现,Atox1在乳腺癌中上调,细胞跟踪显示,在细胞中Atox1沉默后,细胞迁移速度和方向性显著降低。结果表明Atox1通过ATP7A-LOX轴上的协调铜转运体介导乳腺癌细胞的转移。由于单个细胞迁移是乳腺癌转移的早起步骤,肿瘤细胞中的Atox1水平可能是转移潜力的预测指标。所以当细胞内铜含量降低时,可以抑制癌细胞的增殖与转移[22]。
值得一提的是,一些特定类型的癌症更容易受到特定铜依赖过程的影响,例如,限制体内的Cu的水平可以抑制乳腺癌的发展。前文所提到的TM可以将患者血液中的Cu减少一半,且几乎没有严重的副作用。TM治疗降低了血液中骨髓来源的血管生成促进细胞的数量,可能是因为Cu需要激活对这些细胞重要的生长因子。TM对防止肿瘤转移有效,这一作用防止癌症复发作用明显。三阴性乳腺癌是一种恶性程度高且容易复发的乳腺癌,通常会在一年内复发,随后导致较高的短期死亡率。TM治疗可明显提高患者的生存率,有患者在TM治疗8年后没有出现癌症复发的迹象[23]。
4.4 铜与免疫
近期发现,在患各种感染性疾病时,血清铜升高,刺激并增加肝脏合成和释放铜蓝蛋白,用来抵抗微生物的侵袭,而血清铜升高主要与中性粒细胞及巨噬细胞被激活时分泌的一种白细胞内源性物质有关,该物质随血流到远端靶组织发挥重要的免疫调节及杀菌功能。
实验通过给小鼠喂养不同含铜量的饮食,发现低含铜量饮食组的小鼠的器官显示出缺铜的特征性病变时,血清铜、铜蓝蛋白水平降低且免疫反应低下,严重的铜缺乏将损害实验动物的先天和获得性免疫功能。分别用富铜和乏铜饮食喂养小鼠,分离脾淋巴细胞且用单克隆抗体标记后,用流式细胞计分离单个核细胞和决定表型轮廓,结果乏铜鼠的单个核细胞总数,T细胞的绝对数均减少,脾单个核细胞对致有丝分裂抗原的反应率下降,且这种对免疫功能的损害在雄性动物中比雌性动物更加明显[24]。因此维持铜稳态平衡对于机体的健康非常重要。最近有报道通过补充生理剂量铜及其他营养素以改善老年人的免疫反应,取得了有效的结果,且降低感染性疾病的发生率,显示合理补充微量元素可能为免疫功能的恢复提供帮助。
5 结语
铜作为一种重要的微量元素,在人体中发挥着极其重要的作用。铜的内稳态平衡对维持生命体的正常功能非常重要,因此,其摄取与外排过程都受到了严格的调控。铜的稳态被破坏可能导致多种生理功能的改变以及疾病的发生。然而,这一作用也可能被利用进行疾病的治疗,例如通过下调铜的水平抑制癌症的增殖。因此,未来有望可以针对铜相关的疾病,通过铜的配体和配合物的设计,恢复人体内正常的铜水平,实现治疗作用;或针对肿瘤、炎症等病症,通过调控细胞内的铜水平,达到疾病治疗的目的。
猜你喜欢
娃娃乐园·综合智能(2022年9期)2022-08-16
水产科学(2021年4期)2021-07-21
科学大众(2021年9期)2021-07-16
昆明医科大学学报(2020年12期)2021-01-26
食品与生物技术学报(2021年4期)2021-01-17
人工晶体学报(2019年5期)2019-06-18
当代陕西(2019年6期)2019-04-17
分析化学(2018年2期)2018-03-02
中国调味品(2017年2期)2017-03-20
外语学刊(2014年3期)2014-12-03
