
NMDA受体及其拮抗剂的研究进展
2022-05-11 06:54叶玉莹罗扬文于沛
药学研究 2022年4期
叶玉莹,罗扬文,于沛
(暨南大学药学院新药研究所,广东 广州 510632)
作为神经递质中的一种,谷氨酸受体在神经信号传递中扮演着重要的角色,其表达的数量、分布和种类等均影响着正常的神经生理功能,逆转这些受体的功能变化对于治疗或预防神经性疾病有重要意义。兴奋性的突触传导主要通过激活两类谷氨酸受体而实现,即离子型的谷氨酸受体(ionotropic glutamate receptors)和代谢型的谷氨酸受体(metabotropic glutamate receptors),根据对激动剂的亲和力不同,分为3个亚型N-甲基-D-天冬氨酸(NMDA)受体、α-氨基-3-羟基-5-甲基-4-异亚唑丙酸(AMPA)受体和Kainate受体。本文重点关注NMDA受体研究进展,随着研究的深入,以及研究仪器、方法和其他相关领域的发展,NMDA受体的结构逐渐明确,NMDA受体有多种亚型,由不同亚单位组成的受体亚型,具有不同的生物物理和生物化学特性。本文将从结构分布和生理活性这两方面详细总结NMDA受体各亚型的特点,介绍并汇总了目前研究较多的NMDA受体拮抗剂,综合文献的研究,提供该受体在神经性疾病中的重要作用,为相关研究提供信息依据。
1 NMDA受体的结构和分布
NMDA受体分布在全脑中,以海马、大脑皮质、纹状体、杏仁体为主。目前已鉴定出了多种NMDA受体亚基,包括广泛表达的NR1,4个不同的NR2亚基(A、B、C、D),两个NR3子单元(A和B)。
1.1 NR1 NR1是NMDA受体的基本亚基,是NMDA受体复合物的功能性亚单位,是实现该受体离子通道的功能所必需的,且NR1形成离子通道,是调节能力最强的神经递质受体,广泛地分布在中枢神经系统。
1.2 NR2 NR2是多基因家族,分别编码为NR2A、NR2B、NR2C、NR2D。其中NR2A和NR2B对NMDA受体的结构和功能十分重要。含NR2A或NR2B的NMDA受体有突触后密集区蛋白(PSD-95)等[1]一些相同的结合配偶体,NR2A可以与Homer蛋白、β-Catenin蛋白[2]和Rab亲和蛋白3A结合[3],在成人脑部中主要表达在突触内[4];而NR2B则与突触RasGTP酶激活蛋白(SynGAP)等结合,在成人脑部中表达在突触外[5],两者较其他NMDA受体亚型都有较大的单通道电导系数,对胞外镁离子的阻断更敏感,钙离子渗透率更大[6]。
NR2亚单位分布不同,且在成长过程中也会变化。在胚胎时期,NR2B和NR2D是主要的亚单位,前者表达于中枢神经系统中,后者只表达在间脑和脑干;出生两周后,NR2A在中枢神经系统的表达逐渐增多,NR2B的表达在出生后7~10天达到高峰并限制在前脑区域,GluN2C出现较晚且限制在小脑和嗅球中,GluN2D的表达则在出生后发育而下降。NR2B与NR2A有着代偿的联系,减少突触NR2B的表达可使NR2A的表达增加[7],且抑制因子-1沉默转录因子(REST)参与了NR2B向着NR2A随年龄增大而成熟的转变[8]。
1.3 NR3 NR3主要在发育中的中枢神经系统中表达,NR3经过不同的剪接得到两个成员:NR3A和NR3B。NR3A在胚胎时期含量较低,但出生后很快升高,在青春期减少,主要分布于海马、皮质和丘脑等;NR3B主要分布于脑干和脊髓的躯体运动神经元,NR3单独不能形成功能受体,但是NR3可以与NR1和NR2形成NMDA受体复合物,起到负性调节的作用。
1.4 NMDA受体异四聚体的组成 功能性的NMDA受体是一个由两个必需的NR1亚基和两个NR2亚基或NR3亚基构成的异四聚体(见图1),这些亚基结构高度相似,进而构成胞外的氨基末端域(amino-terminal domain, ATD)、胞外的配体结合域(ligand binding domain,LBD)、跨膜区(transmembrane domain,MD)和胞内羧基末端域(carboxy-terminal domain,CTD),与ATD相连的LBD进而与MD连接形成离子通道,MD的螺旋结构与CTD相连[9]。LBD由S1和S2两个子结构域,其中在上部的S1结构具有一定刚性并与ATD相连,而在下部的S2结构具有一定的可变范围并与MD相连,LBD与MD相连的可变性对于形成NMDA受体离子通道结构有着重要的作用。MD有3个跨膜的螺旋结构M1、M3和M4以及成孔凹环的M2, M3在谷氨酸门控型离子通道中有着最保守的片段,有关于与NMDA受体相似的AMPA受体的研究推测MD的打开是通过M3的自转或远离M2中心的侧向位移而完成[10];M2尖端有一个关键的QRN位点决定了钙离子对通道的渗透性。NR2A或NR2B的CTD有很多可以影响到NMDA受体活性的蛋白质相互作用和磷酸化位点,鼠海马神经元NR2A的CTD中的羟基端与PSD-95的相互作用介导着NMDA受体的聚集分布[11]。

图1 GluN1A/GluN2B NMDA 受体离子通道的晶体结构
可以看出,NMDA受体的结构和分布整体表现出的特点与其激活或抑制的状态以及生理活性有关。NMDA受体结构上的不同结合位点、不同亚基组成的亚型不同在分布上有各自的特点,它们有各自的时空变化特点,存在独特和交叉的部分,提示与个体生长发育过程各种生理功能的成熟有关。
2 NMDA受体的生理活性
亚基不同的NMDA受体激活后产生的生理活性有差异。
2.1 GluN1亚基的生理活性 GluN1亚基是所有NMDARs的重要组成部分,与NR2和/或NR3的两个亚基组成NMDA受体通道。此外,GluN1亚基上存在甘氨酸 (Gly) 结合位点,调节NMDA受体的激活。GluN1亚基也与神经元细胞死亡有关,有文献报道GluN1亚基氮-末端域(N-terminal domain,NTD)的一种配体,即组织型纤溶酶原激活剂(tissue-type plasminogen activator,tPA)调整着GluN1-NMDARs动力学从而控制着神经元的死亡[12], Castillo-Gómez等[13]研究发现针对GluN1-NMDARs的自身抗体存在致病潜力。
2.2 GluN2亚基的生理活性 NR2亚基分为NR2A、NR2B、NR2C和NR2D4四种。NR2A调节着神经元NMDA受体诱导的小神经胶质细胞与神经元细胞的物理相互作用[14],减少齿状颗粒神经元中NR2A-NMDARs的表达,显著抑制树突生长[15],甘氨酸通过触发NR2A-NMDARs非离子移变的活性而发挥了神经保护作用[16]。NR2B-NMDARs在神经迁移和皮质分层中扮演着不可缺少的角色,表达在谷氨酸能突触的NR2B-NMDARs直接加速上升途径突触的细化[17],其活性的正反馈对于青少年学习过程的视觉记忆有着启动的作用[18],在agouti相关肽神经元中参与了对体重平衡和血糖平衡的中央控制[19]。至于NR2C-NMDARs则在局部缺血后介导着神经保护作用[20],NR2C敲除模型小鼠表现出精神分裂症样的异常,如认知障碍和前脉冲抑制缺陷,在氯胺酮诱导的行为敏感性的维持上有重要作用[21],与NR2B-NMDARs一起促成丘脑底核中突触的活性。另外突触前包含NR2B、NR2C和NR2D的NMDA受体在孤束核可能控制着迷走神经的传入兴奋性。
2.3 GluN3亚基的生理活性 GluN3亚基有GluN3A和GluN3B两种亚型。在亨廷顿氏舞蹈病动物模型中,GluN3A通过增强突触传导而促进NMDA生成[22],表达在嗅觉系统的GluN3A与嗅觉系统的发育有关[23]。NR3A在早期发育期间在CNS中广泛表达[24],而NR3B在成人的运动神经元群体中富集[25]。
由此可见,亚型不同的NMDA受体的活性存在着交叉和差异,这是NMDA受体成为治疗神经性疾病靶点的一部分困难所在。NMDA受体的过度激活会导致神经系统中突触功能发生改变,进而引起中风、外伤性脑损伤、亨廷顿氏舞蹈病、阿尔兹海默病、精神分裂症和抑郁症等的发生,抑制NMDA受体的活性可以减轻兴奋毒性,预防和减缓神经元的损伤。
3 NMDA受体拮抗剂
NMDA受体具有5个不同的结合位点,分别为①递质结合位点 ;② 甘氨酸调节位点;③离子通道孔结合位点;④ 多胺调节位点;⑤ Zn2+结合位点。根据结合位点的不同,分为不同的靶向NMDA受体的药物,下面主要介绍靶向NMDA受体甘氨酸位点、多胺调节位点及离子通道孔结合位点的药物。
3.1 甘氨酸位点 甘氨酸在可以作为NMDA受体必不可少的共同配体,结合到NMDA受体甘氨酸结合位点上,促进NMDA受体的活性,甘氨酸也可以直接激活NR3-NMDA受体,具有兴奋性递质的功能。
3.1.1 L-701,324 L-701,324(2-氯-1-羟基-7-苯氧基苯基喹啉酮)是一种有效的NMDA拮抗剂,通过阻断其甘氨酸B结合位点来拮抗NMDA受体的活性(结构式见图2)。L-701,324可用于缓解焦虑、紧张和焦虑障碍、促进镇静,用于预防癫痫发作或降低其严重程度的药物,L-701,324在小鼠中具有抗抑郁药样活性,部分是通过促进海马BDNF系统介导的[26]。
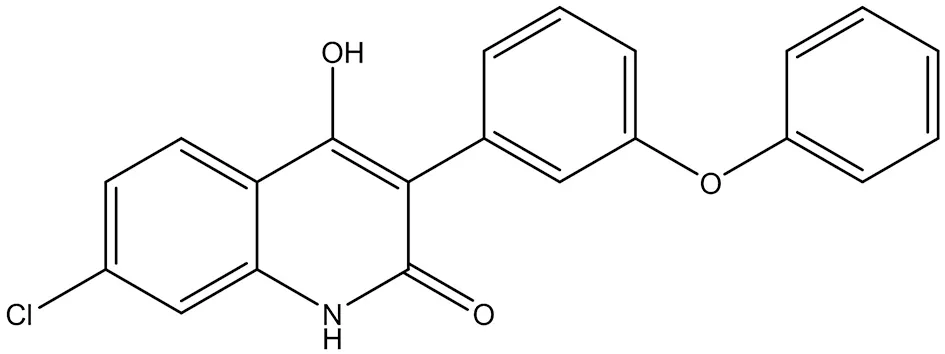
图2 L-701,324结构式
3.1.2 ACEA-1021 ACEA-1021(6,7-二氯-5-硝基-1,4-二氢喹喔啉-2,3-二酮)与具有纳米摩尔亲和力的NMDA受体的甘氨酸位点结合,并且对非NMDA(AMPA / kainate)受体表现出相对较少的亲和力[27](结构式见图3)。在癫痫发作之前或之后立即用ACEA-1021治疗后,可以防止高达86%接受致命剂量可卡因的小鼠死亡[28]。

图3 ACEA-1021结构式
3.1.3 GLYX-13(Rapastinel) GLYX-13 是一种N-甲基-D-天冬氨酸受体 (NMDAR) 甘氨酸位点功能部分激动剂和认知增强剂,也显示出快速的抗抑郁活性,无精神分裂副作用(结构式见图4)。用于治疗重度抑郁症(NCT03614156,NCT03560518),GLYX-13通过增强导水管周围灰质中的AMPA受体功能来缓解慢性应激诱导的抑郁样行为[29]。

图4 GLYX-13结构式
3.1.4 AV-101 AV-101(2-氨基-4-(2-氨基-4-氯苯基)-4-氧代丁酸) 是NMDA受体GlyB位点的选择性拮抗剂(结构式见图5),在双盲随机并有安慰剂对照的Ⅰ期临床试验中显示出其安全性高和药物动力学特点良好,可以用于治疗神经性疼痛甚至是抑郁症[30]。
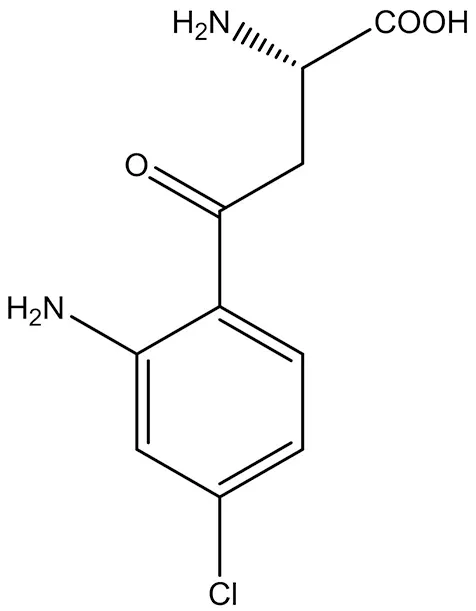
图5 AV-101结构式
3.1.5 D-环丝氨酸(D-Cycloserine) D-环丝氨酸(4-氨基-1,2-恶唑烷-3-酮)是NMDA受体的共激动剂(结构式见图6),在临床上对精神分裂症患者的神经可塑性没有影响,在LTP测试中表现出很大的前高频视觉刺激神经反应,说明D-环丝氨酸能结合NMDA受体[31],且在仍未结束的一个临床试验中被用于治疗抑郁症(NCT03062150)。
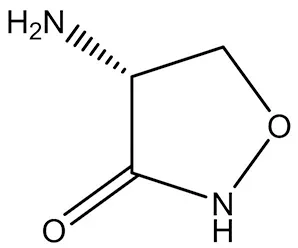
图6 D-环丝氨酸结构式
3.2 多胺结合位点 广谱的NMDA受体拮抗剂能影响所有NMDA受体而产生严重的精神副作用 ,限制了其临床运用,因此,选择性作用NR2B的NMDA受体拮抗剂成为更安全、有效的药物。
3.2.1 MK-0657 (CERC-301) MK-0657 (4-甲基苄基(3S,4R)-3-氟-4‐[(嘧啶-2-ylamino)甲基]哌啶-1-羧酸酯)是一种口服生物可利用的选择性N-甲基-D-天冬氨酸(NMDA)受体亚基2B(GluN2B)拮抗剂(结构式见图7),目前正处于Ⅱ期临床试验中(NCT01941043,NCT02459236),其抗抑郁作用的工作机制尚不清楚,Lei等[32]研究发现MK-0657缓解了慢性约束应激(CRS)诱导的小鼠外侧缰中的绝望样行为,这种缓解可能涉及LHb中BDNF表达的降低,从而降低神经元活性。
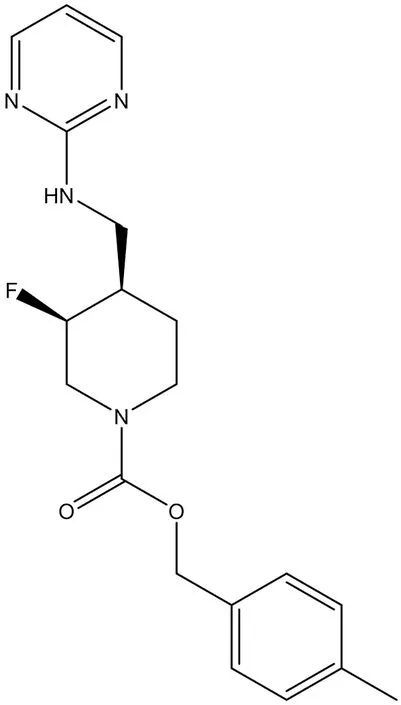
图7 MK-0657结构式
3.2.2 PEAQX PEAQX ([[[1S)-1-(4-溴苯基)乙基]氨基]-(2,3-二氧代-1,4-二氢奎噁啉-5-基)甲基]膦酸)是一种选择性GluN2A拮抗剂(结构式见图8),可用于治疗皮质播散性抑郁症[33]及精神分裂症[34]。Mares等[35]的研究结果表明,GluN1/GluN2A首选拮抗剂PEAQX的抗惊厥作用具有年龄依赖性差异。
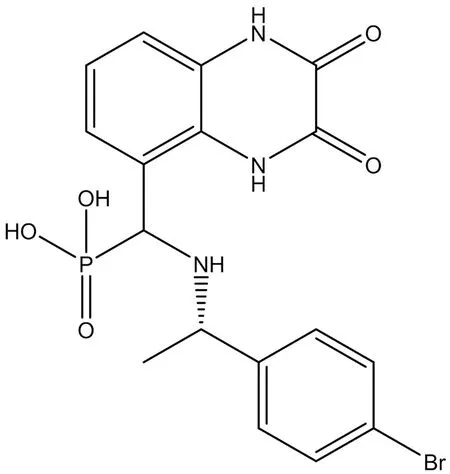
图8 PEAQX结构式
3.2.3 艾芬地尔(ifenprodil) 艾芬地尔(4-[2-(4-苄基哌啶-1-基)-1-羟丙基]苯酚)是一种口服生物可利用的N-甲基-D-天冬氨酸(NMDA)受体拮抗剂(结构式见图9),用作脑血管扩张剂[36],并在临床试验中用于治疗药物成瘾[37],特发性肺纤维化和COVID-19。Ifenprodil结合并抑制谷氨酸NMDA受体GluN2B,从而防止NMDAR信号传导。抑制了神经元的兴奋性毒性,从而潜在地增强了认知功能。Ifenprodil可快速改善抑郁样行为,激活mTOR信号传导并调节CUMS大鼠海马体中的促炎细胞因子[38]。一项关于ifenprodil治疗COVID-19确诊住院患者的安全性和有效性的研究正在进行2b/3期临床试验(NCT04382924)。
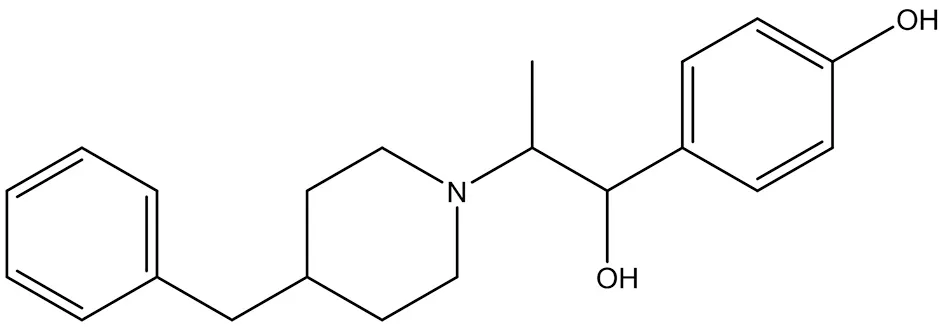
图9 艾芬地尔结构式
3.3 离子通道孔位点 非竞争性NMDA受体拮抗剂能与NMDAR离子通道孔深部的PCP位点结合,阻断与NMDAR耦联的钙通道, 减少Ca2+内流,抑制NMDAR的受体-通道的活动。目前发现的作用于NMDAR离子通道孔位点的药物主要包括:苯环己哌啶 (phencyclidine, PCP) 、地卓西平 (dizocipine, MK-801) 、氯胺酮 (ketamine)、美金刚 (memantine) 、拉尼西明 (lanicemine, AZD6765) 、氧化亚氮 (nitrous oxide,N2O)。
3.3.1 Dizocipine(MK-801) MK-801是NMDA受体(受体,N-甲基-D-天冬氨酸)的强效非竞争性拮抗剂(结构式见图10),影响认知功能、学习和记忆。它具有NMDA受体拮抗剂,麻醉剂,抗惊厥药,烟碱拮抗剂和神经保护剂的作用。由于其严重的精神副作用,如幻觉、妄想、言语贫乏、意志减退等,禁用于临床,其使用主要限于动物和组织实验[39]。

图10 MK-801结构式
3.3.2 氯胺酮(ketamine) 在细胞实验中,氯胺酮(结构式见图11)通过抑制PKC/ERK通路而引起海马神经元的凋亡可以被兴奋性的NMDA受体激活所反转[40]。氯胺酮表现出快速的降低抑郁症患者情绪低沉程度的效果,这种效果可能是基于其改变额顶骨连接模式的能力[41],并且其代谢产物2R,6R-hydroxynorketamine对AMPA受体有兴奋活性和抗抑郁的药理活性,值得注意的是,这个代谢产物可能表现出更少的氯胺酮相关副作用[42];但是其对NMDA受体的抑制能力却弱于氯胺酮,似乎2R,6R-hydroxynorketamine的抗抑郁作用不是完全由于其抑制NMDA受体的活性[43]。最近的一项实验显示,氯胺酮诱导催眠效果和神经可塑性是通过破坏磷酸化MAPK激酶(p-MEK)与磷酸化p-ERK的偶联,下调p-ERK水平并上调磷酸化Fas相关死亡域蛋白(phosphorylated Fas-associated with death domain protein,p-FADD)水平[44]。氯胺酮作为经典的NMDA受体拮抗剂,曾经在临床试验中用于术后止痛(NCT02950233)[45]、重度抑郁症(NCT03609190)[46],现在也有用于耳鸣(NCT03336398)、酒精复发(NCT02649231)和难治性抑郁症(NCT02782104)的临床试验正在进行。
图11 氯胺酮结构式
3.4 其他NMDA受体相关药物
3.4.1 右美沙芬(dextromethorphan) 右美沙芬是非竞争性的NMDA受体拮抗剂(结构式见图12),由Roche 公司开发,曾在临床试验中用于抑郁症(NCT02860962,NCT02153502)和精神分裂症(NCT02477670),现有正在进行的临床试验用于治疗化疗所致外周神经病变(NCT02271893)、亨廷顿病(NCT03854019)和痴呆型激动症(NCT02446132),临床上主要是用于镇咳。研究显示,右美沙芬对血管性痴呆(vascular dementia)大鼠的海马神经损伤和认知能力缺陷有预防作用[47],但由于其与5-HT受体的作用可能导致5-羟色胺综合征,会出现呕吐、恶心、腹泻和嗜睡等副作用[48]。
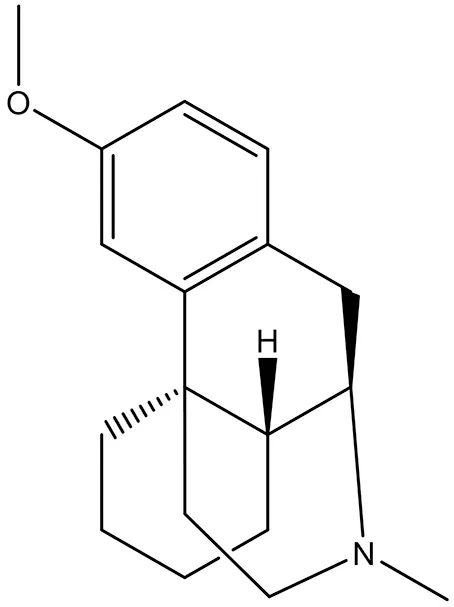
图12 右美沙芬结构式
3.4.2 金刚烷胺(amantadine) 金刚烷胺(结构如图13所示)也是非竞争性的NMDA受体拮抗剂,曾被用于治疗PD、药物导致的锥体束外反应以及病毒感染病等,在人体中可能导致反副交感神经生理样副作用(如口干、尿潴留、便秘、恶心、头晕和失眠等)。最近的研究显示[49],金刚烷胺增强大鼠运动和探寻活动相关的黑质纹状体和中脑缘的多巴胺功能,在一项随机双盲试验中[50],Nourbakhsh等[51]发现金刚烷胺改善多发性硬化疲劳方面并不优于安慰剂,并导致更频繁的不良事件。

图13 金刚烷胺结构式
3.4.3 石杉碱A(huperzine A) 石杉碱A(结构如图14所示)为蛇足石杉(Huperziaserrata)中成分,已批准用于治疗AD,是一个选择性的AChE拮抗剂和非选择性的NMDA受体拮抗剂,有抗炎、镇痛和抗痉挛作用[52],陈庆状等[53]研究发现HupA可通过减少Aβ与淀粉样蛋白结合醇脱氢酶(ABAD)的结合而改善线粒体损伤,进而改善AD小鼠的认知和记忆功能障碍。曾在临床试验中用于精神分裂症(NCT00963846)和痴呆症(NCT01012830),也有用于外伤性脑损伤(NCT01676311)和提高认知能力(NCT01676311)的临床试验正在进行,另外一项实验证明饮食诱导的肥胖小鼠中,Hup A治疗可以有效地改善认知功能[54]。

图14 石杉碱A结构式
现将与NMDA受体有关的药物总结如表1所示。

表1 NMDA受体相关药物
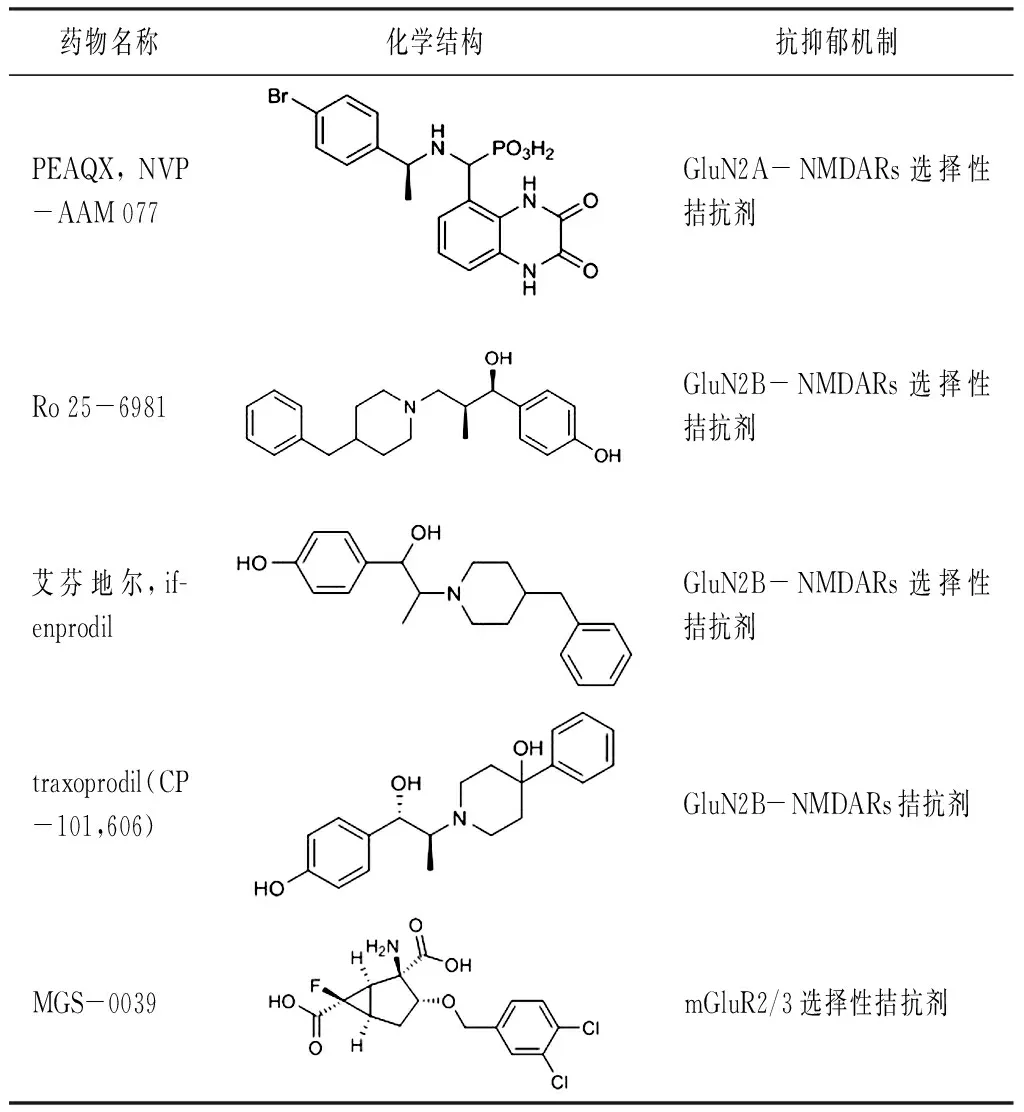
表1(续)
4 总结与展望
至此,NMDA受体在神经性疾病中的重要位置已不言而喻,其作为治疗和预防神经性疾病的靶点的潜力大,但由于分型的高度同源和其广泛的生理活性,单一化合物针对性成药较难,温和的、亚型选择性强的NMDA受体调节剂又或联用共激动剂和变构调节剂有着更好的成药前景,由回顾前人的研究成果与已经上市的有关NMDA受体的药物可见。因此,接下来的研究方向便可能为:①已有NMDA受体拮抗剂的结构改造,以增加其对不同位置、不同亚型的NMDA受体的选择性;②寻找更具选择性的变构调节剂或共激动剂;③通过基础研究构建与谷氨酸能神经生理活动相关的生理信号系统,通过间接的靶向其他重要靶点以影响NMDA受体和有关的生理信号通路,以达到调节谷氨酸能神经功能的目的;④进一步研究药物相互作用,寻找在治疗作用和副作用上互补或协同的药物组合,以达到系统地调节NMDA受体活性的目的。
我们有理由相信,随着更多的研究成果的浮出,关于NMDA受体的探索将会在神经领域中继续深入,治疗和预防神经性疾病的研究道路由此开辟。
猜你喜欢
新疆农业科学(2022年6期)2022-07-13
麦类作物学报(2022年3期)2022-05-19
临床与实验病理学杂志(2022年3期)2022-04-06
中学生数理化(高中版.高考数学)(2021年10期)2021-12-02
中学生数理化·高三版(2021年10期)2021-11-01
科学之谜(2018年9期)2018-12-17
中国医药导报(2017年34期)2018-01-29
试题与研究·高考理综生物(2016年4期)2017-03-28
中学生数理化·高三版(2016年12期)2017-03-02
中学生数理化·高三版(2016年12期)2017-03-02
