
基于D(-)/L(+)-对羟基苯甘氨酸配体的镉配合物的合成、结构及荧光识别性能
2022-05-10 11:00李思浓赵美红宋会花
河北师范大学学报(自然科学版) 2022年3期
李思浓,赵美红,宋会花
(河北师范大学 化学与材料科学学院,河北 石家庄 050024)
手性配位聚合物(CPs)因其在不对称催化、非线性光学材料、手性分离和识别、荧光等领域中的潜在应用,引起了科研人员的极大兴趣[1-7].目前手性配位化合物的合成方法主要有3种:1)利用手性配体为构筑单元与金属离子的自组装;2)使用手性模板、手性溶剂或催化剂从非手性构建块诱导;3)利用非手性前体进行自发结晶拆分.其中利用手性配体的自组装是最直接有效的方法[8-10].然而,在自组装合成配合物的过程中,金属离子、配体、体系pH值、金属与配体的比例、反应温度、溶剂等因素都影响着产物的结构[11-13].因此,控制合成期望的手性配位聚合物仍是一项挑战性工作.
手性配体的选择是合成结构新颖且性质独特的手性配合物的关键.氨基酸及其衍生物廉价易得,含有丰富的N,O原子,不仅具有多样的配位模式(单齿、螯合、桥联),还可与软硬不同的金属离子配位.此外,氨基氮原子、羰基氧原子、羟基氧原子可以充当氢键的供体和受体,通过氢键作用能够使小分子配合物形成维数更高、更稳定的超分子结构[14-15].D/L-对羟基苯甘氨酸(D/L-Hhpg)作为氨基酸衍生物,具有良好的配位性能,容易获取且无毒无害,可作为合成手性配合物的理想配体.目前以其为手性配体合成的配合物还非常有限,2004年,Mukhopadhyay等[16]报道了一例金属铜配合物[Cu(4hpg)(bpy)]·2H2O(bpy = 2,2′-联吡啶,hpg=D-4羟基苯甘氨酸);2015年,Yao课题组[17]报道了一例金属铜配合物[Cu(hpg)2(H2O)]n.本课题组已成功合成了几例锌和铜的手性配合物,并对其荧光识别性质及电化学性质进行了探究[18-20];但是,基于D/L-Hhpg的新型配位聚合物还有待于进一步的开发.
在废水、土壤甚至食品中均发现一些有毒的金属离子和有机小分子,这些毒物对人类健康和环境安全将产生极大威胁,所以对有害化学物质的快速检测非常有必要.为了设计出具有高灵敏度荧光响应的配合物,常选择以Zn(Ⅱ)和Cd(Ⅱ)这些具有d10的闭壳层结构的金属离子,这些离子在荧光性能上表现出明显的优势[21-24].现有的用于检测Cu2+和Fe3+的d10过渡金属配合物为数不多,并且大多数都在有机溶剂中进行[25].因此,设计出简单易合成、绿色环保、成本低并可用于在水相中检测Cu2+和Fe3+的d10过渡金属有机配合物很有意义.另外,研究发现含氮辅助配体在设计与构造结构多变的配位聚合物方面扮演着重要角色,如1,2-二(4-吡啶)乙烷、邻菲罗啉、喹啉、4,4′-联吡啶等,可以提供热稳定性良好的框架,诱发微妙的环境变化,可能会改变最终结构等,因此常常在自组装过程中被用来稳定超分子结构[26].
基于以上考虑,本文中,笔者以D/L-Hhpg作为手性配体,1,3-双(4-吡啶基)丙烷(bpp)为辅助配体与金属镉盐(Cd(NO3)2·6H2O)自组装合成了一对手性配合物:{[Cd(D-hpg)(bpp)(H2O)]·(NO3)·(H2O)}n(1),{[Cd(L-hpg)(bpp)(H2O)]·(NO3)·(H2O)}n(2),通过单晶X射线衍射(SCXRD)、元素分析(EA)、红外光谱(IR)、粉末X射线衍射(PXRD)、热重分析(TGA)及圆二色谱(CD)对其结构进行了分析表征.此外,研究了配合物的固体荧光性质及对Cu2+,Fe3+的荧光识别性能.
1 实验部分
1.1 仪器及试剂
红外图谱用日本岛津FTIR-8900型红外光谱仪测定;元素分析用德国Elementar公司的Vario EL型 C,H,N元素分析仪测定;热重分析用德国耐驰公司的 NETZSCH STA 449 F3同步热分析仪测定;X射线单晶衍射测定在德国Bruker公司Smart-CCD单晶衍射仪上进行.
所用试剂均为市场上购买的分析纯试剂.
1.2 配合物1和2的合成
称取Cd(NO3)2·6H2O 0.061 6 g(0.2 mmol),D-Hhpg 0.016 7 g (0.1 mmol)置于洁净的烧杯中,向其中加入8 mL蒸馏水,搅拌溶解,将溶有bpp 0.019 8 g(0.1 mmol)的8 mL乙醇溶液缓慢加入上述溶液,用1 mol/L NaOH调节溶液pH=6.4,并在室温下继续搅拌25 min.过滤,将滤液室温静置10 d左右,得到无色棒状晶体,产率68 % (以Cd计).元素分析C21H26N4O8Cd (1,Mr=574.86) 实验值(%,理论值):C 43.95(43.84),H 3.49(3.52),N 9.68(9.74).IR主要吸收峰值(KBr,cm-1)ν:3 327(s),3 271(w),1 618(m),1 604(s),1 597(w),1 550(s),1 518(m),1 504(w),1 423(w),1 383(s),1 367(s),1 267(m),1 232(m),1 174(s),1 120(m),1 016(s),1 008(m),931(w),829(s),773(m),596(w),513(w),497(w).
配合物2的合成方法同1,区别是用L-Hhpg替代D-Hhpg.配合物2为无色棒状晶体,产率64 % (以Cd计).元素分析C21H26N4O8Cd (2,Mr=574.86)实验值(%,理论值):C 44.06(43.84),H 3.35(3.52),N 9.71(9.74).IR主要吸收峰值(KBr,cm-1)ν:3 327(s),3 271(w),1 618(m),1 604(s),1 597(w),1 550(s),1 518(m),1 504(w),1 423(w),1 383(s),1 367(s),1 267(m),1 232(m),1 174(s),1 120(m),1 016(s),1 008(m),931(w),829(s),773(m),596(w),513(w),497(w).
1.3 晶体结构的测试
在298 K时,利用SMART-CCD单晶衍射仪测试晶体.石墨单色器处理单色化Mo Kα(λ=0.071 073 nm)射线,选用ψ-ω扫描方法收集数据,F2最小二乘法对非氢原子进行修正[27],采用SHELXTL-97程序对晶体结构进行解析.通过解析配合物1,2获得晶体结构学参数及其相关数据,列于表1.
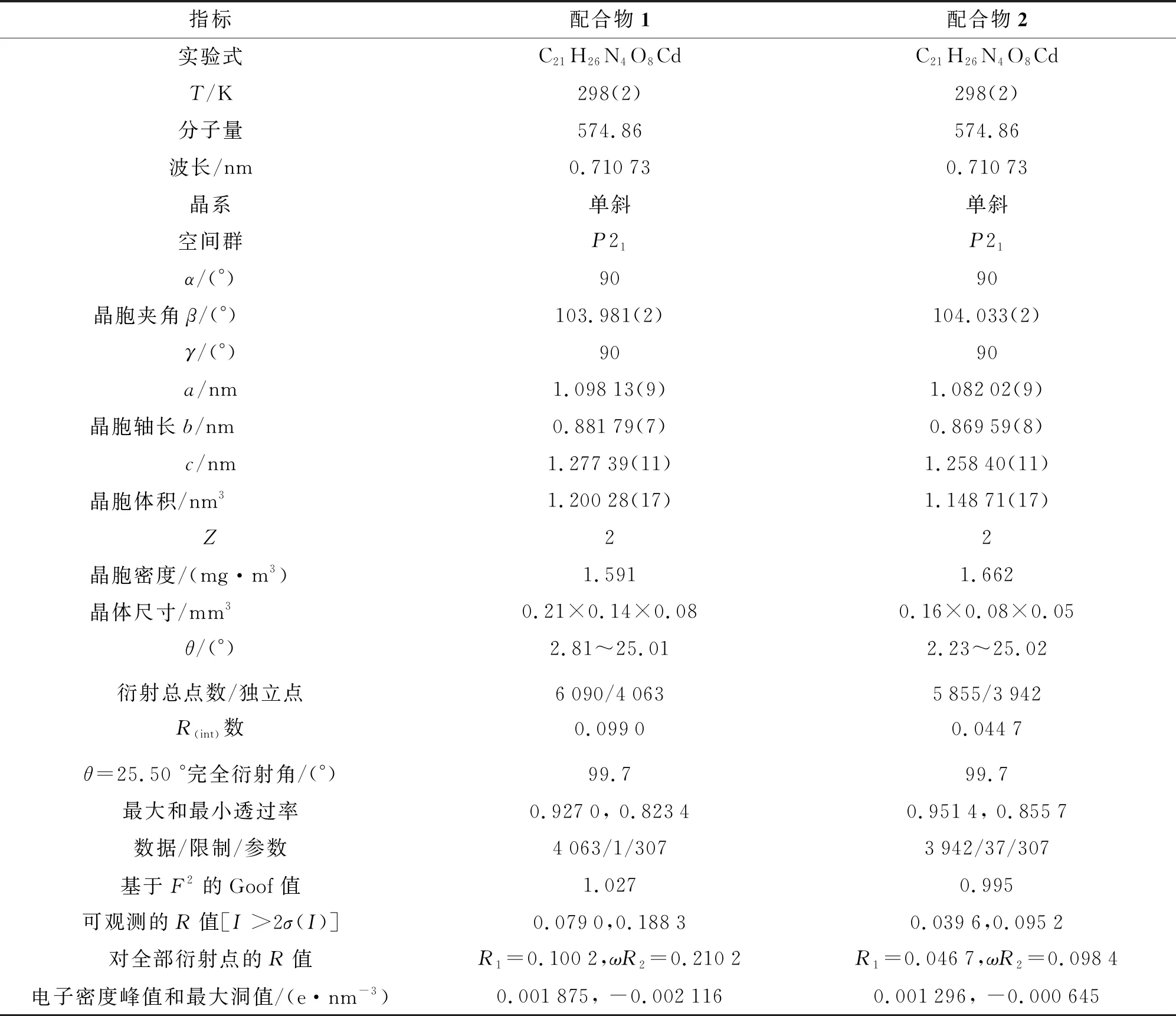
表1 配合物1,2的晶体学结构参数及其相关数据Tab.1 Crystallographic Structure Parameters and Related Data of Compounds 1 and 2
2 结果与讨论
2.1 配合物1,2的晶体结构
由表1可知,配合物1为单斜晶系,P21手性空间群,1D左手螺旋链结构.配合物1的不对称单元是由1个Cd(Ⅱ)阳离子、1个D-hpg-阴离子、1个bpp辅助中性配体分子、1个抗衡阴离子NO3-,1个配位水分子以及1个晶格水分子组成,如图1a所示.配合物的中心Cd(Ⅱ)为六配位,其配位方式为CdO3N3,呈现出变形的八面体构型,O1,O2#1,O4和N1#1处于八面体赤道位置,而N2和N3则处在八面体的轴向位置上.O1,O2,N1分别来自2个D-hpg-配体,N2,N3分别来自2个bpp辅助配体,而O4来自1个配位水分子.Cd—O键长为0.240 2(6)~0.244 9(8) nm,Cd—N键长为0.236 8(9)~0.237 7(10)nm(表2).

图1 配合物1(a)和2(b)的基本结构单元Fig.1 The Basic Structural Unit of Complex 1(a) and 2(b)

表2 配合物1中主要的键长及键角Tab.2 Major Bond Lengths and Boend Angles in Comound 1
配合物1中D-hpg-配体经由羧基氧原子和氨基氮原子以螯合-桥联三齿配位模式连接2个中心金属Cd(Ⅱ),在b轴方向上无限延伸构成1D的左手螺旋链(图2a);而中性配体bpp在b轴方向桥连2个相邻的中心金属Cd(Ⅱ),因此配合物1仅为1D链结构.配合物1中有游离的NO3-抗衡阴离子、晶格水分子、配位水分子,D-hpg-配体中氨基氮以及羧基氧原子之间存在大量的氢键作用(N(1)-H(1A)…O(3)=0.308 9 nm,N(1)-H(1B)…O(5)#5=0.305 7 nm,O(3)-H(3)…O(8)#3=0.278 7 nm,O(4)-H(4B)…O(8)#6=0.282 9 nm,O(4)-H(4C)…O(8)#7=0.298 5 nm,O(4)-H(8C)…O(6)#3=0.284 6 nm;#3:x,y,z+1/2;#5:-x+1,y+1/2,-z+3;#6:x,y+1,z;#7:-x+1,y+1/2,-z+2).通过这些氢键作用,配合物1最终形成3D超分子结构,如图3a所示.
将配合物1,2的晶体数据及图1a,b进行对照可知,配合物1和2互为一对对映异构体.2例配合物的区别在于配合物2中L-hpg-配体以螯合-桥联三齿配位模式连接相邻的2个中心金属Cd(Ⅱ),在b方向上无限延伸构成1D右手螺旋链(图2b).同样的,配合物2中的中性配体bpp在b轴方向桥连2个相邻的中心金属Cd(Ⅱ),因此配合物2仅为1D链结构.其中,Cd—O键长在0.237 4(3)~0.262 4(4) nm,Cd—N键长在0.234 0(5)~0.235 1(10) nm(表3).配合物2中有游离的NO3-抗衡阴离子、晶格水分子、配位水分子,L-hpg-配体中氨基氮以及羧基氧原子之间存在大量的氢键作用(N(1)-H(1A)…O(6)#1=0.301 9 nm,N(1)-H(1B)…O(3)#6=0.308 5 nm,O(3)-H(3)…O(8)#7=0.275 0 nm,O(4)-H(4B)…O(8)#4=0.295 4 nm,O(4)-H(4C)…O(1)#2=0.280 1 nm,O(8)-H(8C)…O(7)=0.285 4 nm,O(8)-H(8D)…O(5)=0.279 1 nm;#1:x+1,y+1,z;#2:-x+2,y+1/2,-z+1;#4:-x+1,y+1/2,-z+1;#6:-x+2,y+1/2,-z+3;#7:-x,y+1/2,-z+1),使得配合物2形成3D超分子结构,如图3b所示.

图2 配合物1中的左手螺旋链(a)和配合物2中的右手螺旋链(b)Fig.2 Left-hand Helical Chain in Compound 1 (a)and Right-hand Helical Chain in Comound 2(b)

表3 配合物2中主要的键长及键角Tab.3 Major Bond Lengths and Bond Angles in Compound 2

图3 配合物1(a)和2(b)的3D超分子结构Fig.3 3D Supermolecular Structures of Compounds 1(a) and 2(b)
2.2 配合物1和2的表征
2.2.1 配合物1和2的红外光谱

图4 配合物1和2的红外光谱Fig.4 IR Spectra of Compounds 1 and 2

2.2.2 配合物1和2的PXRD图谱
室温条件下、5 °≤ 2θ≤ 50 °,测得了配合物1和2的X射线粉末衍射谱图(见图5),可以看出,配合物1,2的PXRD实验图谱和理论拟合图谱中主要峰位置是相匹配的,因此所合成的晶体是纯相的.

a.实验图谱;b.理论图谱.图5 配合物1和2的PXRD图谱Fig.5 PXRD Patterns of Compounds 1 and 2
2.2.3 配合物1和2的热重分析
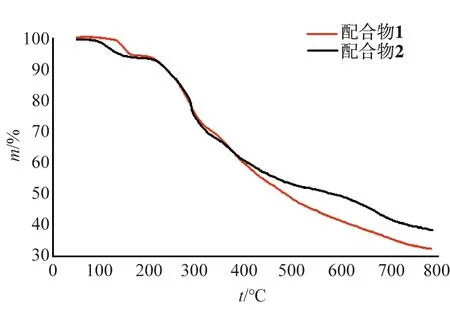
图6 配合物1和2的热重图Fig.6 TGA Curves of Complounds 1 and 2
在N2气氛下,保持20 ℃/min持续升温,测定配合物1,2在25~800 ℃的热稳定性.如图6所示,配合物1在70 ℃之后开始分解,并逐步失重.第1步失重在74.0~184.0 ℃,对应失去1个晶格水分子(实验值5.94 %,理论值6.26 %);之后开始第2步失重,在184.0~330.0 ℃,失重率达23.95 %,分析为配合物主体框架开始部分分解.从330.0 ℃开始第3步失重,并持续失重,直至800 ℃未出现平台,表明在测试温度范围内配合物1并未完全失重.
经分析,配合物2在60 ℃之后开始分解,并逐步失重.第1步失重在65.0~179.0 ℃,对应失去1个晶格水分子(实验值5.89 %,理论值6.26 %);之后开始第2步失重,在179.0~339.0 ℃,失重率达25.57 %,分析为配合物主体框架开始部分分解.从339.0 ℃开始第3步失重,并持续失重,直至800 ℃未出现平台,表明在测试温度范围内配合物2并未完全失重.
2.2.4 配合物1和2的固态荧光性质
室温条件下,测定了配合物1和2的固态荧光图谱,如图7所示.主配体D-Hhpg和L-Hhpg在350~550 nm未出现荧光发射峰.辅助配体bpp激发波长λex=360 nm时,发射波长λem=396 nm;配合物1的λex=340 nm时,λem=402 nm,配合物2的λex=340 nm时,λem=406 nm,对比辅助配体bpp、配合物1和2的荧光图谱可以发现,配合物1和2的荧光发射峰位置都蓝移了20 nm,因此配合物1和2的荧光峰可归属于金属-配体的电荷转移跃迁(LMCT)[29-30].
2.2.5 配合物1和2的圆二色谱分析
室温条件下测定了配合物1和2的圆二色光谱(图8).配合物1在262,288 nm处出现2个负峰,配合物2在261,288 nm处出现2个正峰.根据配合物的CD谱图可知,配合物1和2表现出明显的Cotton效应,并且2个配合物的CD曲线大致呈镜像对称,说明配合物1和2均为纯手性.
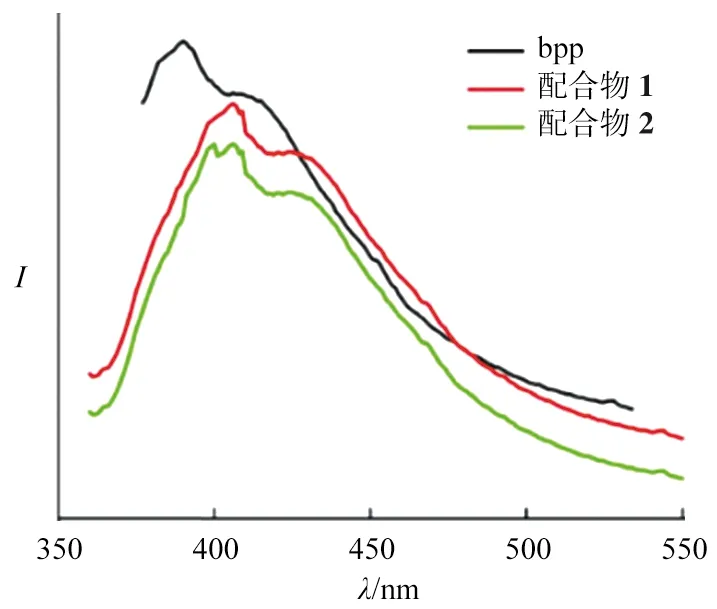
图7 配合物1,2和bpp的荧光光谱Fig.7 Solid Fluorescence Intensities of a bpp and Compounds 1,2

图8 配合物1和2的CD光谱Fig.8 Solid-state CD Spectra of Compounds 1 and 2
2.2.6 配合物1和2对Cu2+和Fe3+的荧光识别
2.2.6.1 配合物1对Cu2+和Fe3+的荧光识别
将一定量的配合物1的水溶液(c=3.2×10-5mol/L)分别加入浓度均为0.01 mol/L的含有Cu2+,Fe3+的硫酸盐水溶液中,以未加入金属硫酸盐的等浓度配合物的水溶液作为参比,在相同条件下测定各溶液的荧光发射谱(图9a),可见,Cu2+和Fe3+对配合物1的荧光有十分明显的猝灭效应.为了更好地解释Cu2+和Fe3+的猝灭作用,测定了上述混合溶液的紫外-可见光谱(图9b),相对于参比溶液,加入Cu2+或Fe3+的水溶液的吸收带完全覆盖了配合物1的吸收带,因此导致了配合物1荧光强度的锐减甚至猝灭现象.为探究配合物1对Cu2+和Fe3+的识别抗干扰能力,向含Cu2+和Fe3+溶液中引入等浓度(0.01 mol/L)的其他金属离子,测其荧光发射谱(图9c,d).由荧光对比图可知,其他金属离子的引入没有干扰到配合物1对Cu2+和Fe3+荧光识别效应.由此可知,配合物1可选择性识别水相中的Cu2+和Fe3+.
2.2.6.2 配合物2对Cu2+和Fe3+的荧光识别
配合物2识别Cu2+和Fe3+的实验方法同配合物1.如图10a所示,Cu2+和Fe3+对配合物2的荧光具有明显的猝灭效应.为了更好地解释Cu2+,Fe3+的猝灭作用,测定了上述溶液的紫外-可见光谱(图10b),引入Cu2+和Fe3+后的配合物紫外吸收带已将作为参比的配合物2的吸收带完全覆盖,因此导致Cu2+和Fe3+对配合物具有荧光一定程度的猝灭效应.为探究配合物2对Cu2+和Fe3+的识别抗干扰能力,向含Cu2+和Fe3+溶液中引入等浓度(0.01 mol/L)的其他金属离子,测其荧光发射谱(图10c,d).由荧光对比图可知,配合物2对Cu2+和Fe3+具有荧光识别能力,且不受其他金属离子的干扰.由此可知,配合物2可选择性识别水相中的Cu2+和Fe3+.
3 结 论
利用D(-)/L(+)-对羟基苯甘氨酸手性主配体,在含N辅助配体bpp的存在下成功合成了一对金属Cd(Ⅱ)的新型对映异构体配合物1,2,配合物分别具有1D左手和1D右手螺旋链结构.配合物1和2均显示出明显的Cotton效应且光谱曲线呈镜像对称关系.配合物1和2具有良好的荧光性能,可用于水溶液中Cu2+和Fe3+的选择性识别.
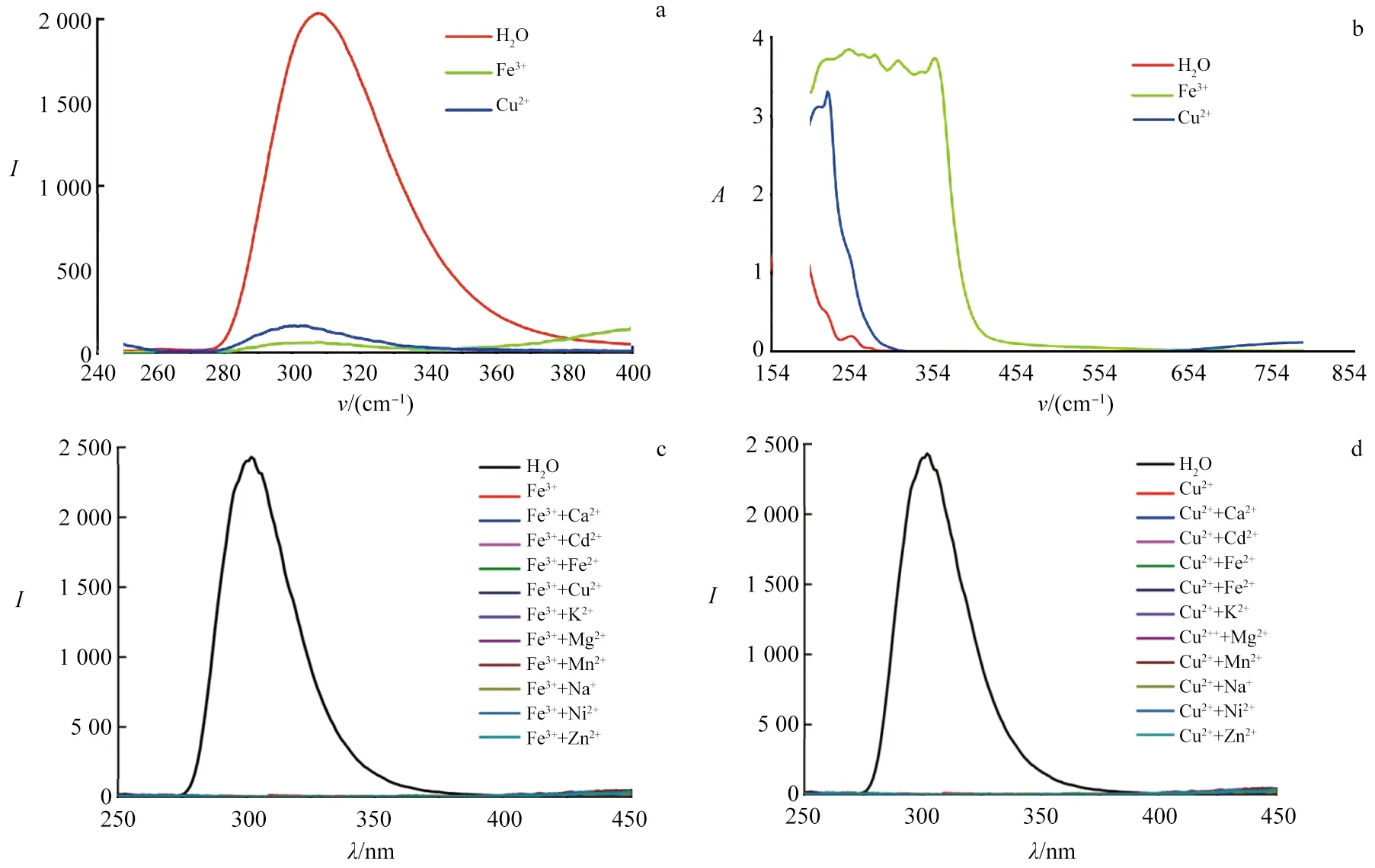
a.引入Cu2+和Fe3+的荧光光谱; b.引入Cu2+和Fe3+的紫外-可见光谱; c.对Fe3+的抗干扰荧光光谱; d.对Cu2+的抗干扰荧光光谱.图9 配合物1对Cu2+和Fe3+的识别图谱Fig.9 Identification Spectra of Compound 1 for Cu2+ and Fe3+
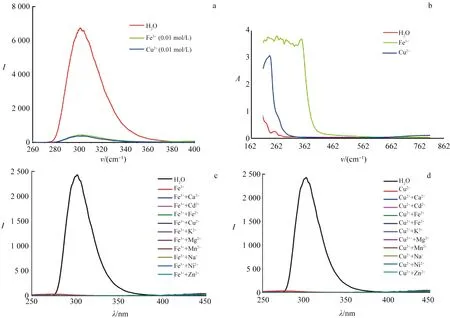
a.引入Cu2+和Fe3+的荧光光谱; b.引入Cu2+和Fe3+的紫外-可见光谱;c.对Cu2+的抗干扰荧光光谱; d.对Fe3+的抗干扰荧光光谱.图10 配合物2对Cu2+和Fe3+的识别图谱Fig.10 Identification Spectra of Compund 2 for Cu2+ and Fe3+
猜你喜欢
分子催化(2022年1期)2022-11-02
科学导报(2022年41期)2022-07-13
原子与分子物理学报(2022年3期)2022-03-05
辽宁科技大学学报(2021年2期)2021-07-22
湖南大学学报·自然科学版(2020年2期)2020-04-17
青岛大学学报(工程技术版)(2019年2期)2019-09-10
人工晶体学报(2019年5期)2019-06-18
当代陕西(2019年6期)2019-04-17
分析化学(2018年2期)2018-03-02
江苏农业科学(2017年20期)2017-11-30
