
基于PBPK模型预测不同晶型利福平的生物等效性
2022-05-09 02:58蔡其霖黎文星尹莉芳
中国药科大学学报 2022年2期
蔡其霖,黎文星,严 真,尹莉芳,*
(1中国药科大学药学院药剂系,南京 210009;2江苏省缓释智能制剂及关键功能性辅料开发与评价工程研究中心,南京 210009)
利福平(rifampicin)作为一种抑制RNA 聚合酶的大环类抗生素,不仅是世界卫生组织推荐的抗结核病一线药物,也是治疗药物敏感性结核病最有效的药物[1]。利福平的用量占抗结核药物总量的四分之一,且被陆续开发出新的临床应用,如治疗阿尔茨海默病等[2]。目前已上市的利福平剂型有片剂、注射剂、胶囊剂、滴眼剂、乳膏剂、口服混悬液等。药物因分子的排列方式不同而表现出不同晶型的情况被称作药物多晶型,利福平结构较复杂,存在多种晶型,目前已发现7 种[3]。药物的晶型不同可能会导致其理化性质不同,也可能影响体内的溶解和吸收情况,从而影响生物利用度。由于忽视药物多晶型导致药物在生产、储存甚至上市后出现生物利用度显著变化的案例并不罕见。例如,氯霉素棕榈酸酯3种不同晶型的生物利用度差异非常显著,就曾发生由于忽视对多晶型的研究导致上市制剂生物利用度显著降低的事故[4]。有文献表明,利福平Ⅰ型与Ⅱ型均属于有效晶型,其中Ⅰ晶型为稳定晶型,Ⅱ晶型为亚稳定晶型[5]。利福平这两种晶型都被开发成上市制剂,本文将研究利福平晶型的差异对生物利用度的影响。
基于生理学的药动学(PBPK)模型是一种建立在药物基本理化性质、制剂的生物药剂学属性、实验个体的生理生化特征等基础上模拟药物在实验个体体内药物动力学过程的模型[6]。PBPK模型的核心就是根据实验个体的生理学特征将其简化为多个参数化的生理房室[7],预测药物在生物体内药动学行为,从而模拟出药物在血浆和组织中的浓度变化情况[8]。建立PBPK 模型的策略在全球众多顶尖制药公司中都得到了应用,FDA 也在药物相互作用指导原则中推荐采用PBPK 建模的方法来预测可能存在的药物相互作用[9]。
GastroPlusTM是美国Simulations Plus 公司研发的一款基于生理学的药动学/药效动力学(PBPK/PD)模拟软件,具有模拟并预测不同制剂在胃肠道各个区域的吸收情况、体内溶出情况和血药浓度-时间曲线[10]等多种功能。本研究运用GastroPlusTM软件,通过建立利福平口服给药的PBPK模型,模拟出给药后的血药浓度-时间曲线,预测药物的吸收情况,分析健康人体口服不同晶型的利福平后的体内药动学行为,通过区别不同晶型的利福平在人体中药动学行为的差异,指导利福平产品的临床应用与制剂开发。
1 材 料
1.1 原料药及试剂
利福平[晶型Ⅰ批号:1812218(M),晶型Ⅱ批号:1811178(M)];布洛芬(山东新华制药股份有限公司);羧甲基纤维素钠[亚什兰(中国)投资有限公司];二甲基亚砜(上海泰坦科技股份有限公司);肝素钠[阿拉丁试剂(上海)有限公司];甲醇、乙腈(美国天地有限公司);甲醇、乙腈为色谱纯,其余试剂为市售分析纯。
1.2 动 物
Wistar 大鼠,体重250 g,雄性,上海西普尔-必凯实验动物有限公司,生产许可证号:SCXK(沪)2018-0006。
1.3 仪 器
GastroPlusTM软 件(Version 9.8,美 国Simula‑tion-Plus 公司);Waters e2695 高效液相色谱仪[沃特世(科技)上海有限公司];D8 ADVANCE 型X 射线衍射仪(德国布鲁克公司);RC806D 溶出试验仪(天大天发科技有限公司);UV1800 紫外分光光度计[岛津企业管理(中国)有限公司)]。
2 方 法
2.1 X射线粉末衍射
使用X 射线衍射仪通过铜靶采集数据,采集条件为40 kV 和40 mA。扫描范围为3~40°(2θ),步长为0.02°(2θ),每步停留时间为1 s。分别对两种晶型的利福平进行扫描,实验结果见图1。
根据扫描结果可知,两种晶型的利福平具有明显不同的特征峰,晶型Ⅰ的特征峰为13.70°和14.40°,晶型Ⅱ的特征峰为9.92°和11.12°,与dePinho Pessoa Nogueira 等[11]研究的图谱特征峰基本一致。两种晶型的利福平均呈现尖锐的衍射峰,表明两者的结晶性均较好。

Figure 1 PXRD patterns of rifampicin form I and II(The position of the arrow indicates the characteristic peak)
2.2 平衡溶解度
利福平在BCS 分类上属于Ⅱ类,具有低溶解度,高渗透性的特点。为了比较晶型Ⅰ和晶型Ⅱ利福平在溶解度性质的差异,选择了不同pH 梯度的介质进行考察。平衡溶解度测定结果如图2所示。

Figure 2 Equilibrium solubility of two crystal forms of rifampicin
根据实验结果,晶型Ⅰ(稳定晶型)与晶型Ⅱ(亚稳定晶型)在溶解度方面特征相似,都表现出明显的pH 依赖性,呈现先减小后增大的趋势。在pH 1.0 介质中溶解度最大,而后溶解度急剧减小,当介质pH 大于4.0时溶解度又逐渐增大。从整体来看,利福平稳定晶型和亚稳定晶型的溶解度无显著差异。
2.3 体外溶出试验
固体制剂在口服后,药物的吸收取决于药物从制剂中溶出或释放的情况、药物在生理条件下的溶解情况及在胃肠道的渗透情况。对于BCS Ⅱ类药物而言,溶出是限制药物吸收的关键步骤。考虑到消化道的生理环境复杂,各部位之间的pH跨度较大,因此选择pH 覆盖范围较广的多种溶出介质测定溶出曲线,能尽可能较全面地反映药物在不同pH 环境下的溶出情况,建立更全面的体内外相关性。
为模拟利福平在体内释放情况,参照“日本药品体外溶出试验信息库”,分别选取pH 1.0盐酸溶液、pH 4.0醋酸盐缓冲溶液、pH 6.8磷酸盐缓冲溶液和水作为溶出介质,进行晶型Ⅰ与晶型Ⅱ利福平体外溶出试验。称取原料药150 mg,将药物粒径控制在75~150 μm,选择桨法,转速50 r/min,介质温度(37 ± 0.5)℃,介质体积900 mL 的条件进行溶出。两种晶型利福平在4 种介质中的溶出结果见图3。
根据体外溶出结果可知,在pH 1.0 盐酸溶液中,1 min 时累积溶出度达到90%,5 min 时基本溶解完全,两种晶型的原料药在该介质中的溶出速率无明显差异,说明两种晶型的利福平都能在酸性的胃部里迅速溶出完全,结合溶解度数据,预测利福平在经过胃部后会以溶液的形式进入肠道。而在其他3种介质中,晶型Ⅱ利福平的溶出速率均快于晶型Ⅰ利福平。这是由于在这3种介质中,利福平的平衡溶解度均较小,两种晶型原料药之间的溶出速率差异得以体现,而这种溶出速率的差异可能是由于不同晶型的极性表面和非极性表面的不同,使得不同晶型利福平被水性介质润湿的难易程度不同,但由于利福平在到达肠道时基本是溶液状态,且大部分利福平口服速释制剂的用法是空腹服用,这3种介质中的溶出速率不再是限制药物吸收的关键步骤。
3 体内研究
3.1 建立大鼠静脉给药PBPK模型
3.1.1 基本信息及静脉给药药动学研究 建立PBPK 模型所用的利福平理化性质参数见表1。利福平理化性质参数主要来源于Drugbank 数据库、发表文献、软件计算值及软件默认值。

Figure 3 Dissolution curves of two crystal forms of rifampicin powder in different medium

Table 1 Parameters required for PBPK model establishment
选取健康的体重约为250 g 雄性Wistar 大鼠6 只,实验期间正常饮食,自由饮水,饲养温度控制在(20 ± 5)℃、饲养湿度控制在30% ~ 70%。配制4 mg/mL 的10%DMSO-生理盐水利福平溶液,按照20 mg/kg的给药剂量对大鼠进行尾静脉注射。
分别于大鼠尾静脉给药后0.16,0.5,1,2,4,6,8,10,12,24 h用毛细玻璃管从眼眶静脉丛取血,血浆样品处理后用高效液相色谱法进行血药浓度测定。
血浆样品处理方法:取血浆样品100 μL,置于离心管中,加入2 mg/mL 布洛芬内标液20 μL 及乙腈250 μL,涡旋混匀,15 000 r/min 条件下离心,取上清液作为供试品溶液,用于高效液相色谱分析。
色谱柱选择C18柱(4.6 mm× 150 mm,5 μm),流动相为磷酸盐缓冲液(pH 5.8)-乙腈-甲醇(30∶20∶50),柱温为40 ℃,检测波长为254 nm,流速为1.0 mL/min,进样量为20 μL。
最终测定的静脉给药后血药浓度-时间曲线见图4。
3.1.2 建立大鼠静脉给药PBPK 模型 首先在GastroPlusTM软件中输入利福平相应的理化参数及生物药剂学参数,血浆占全血百分比取平均值55%,血浆蛋白结合率参考DrugBank 数据库取89%。选择生理学模型为大鼠,再向PKPlus 模块中导入大鼠静脉注射利福平后的血药浓度数据,采用房室模型法和非房室模型法计算各房室的药动学参数,通过比较1/Yhat^2权重的大小来确定最佳的模型,以此建立大鼠静脉给药的PBPK模型。
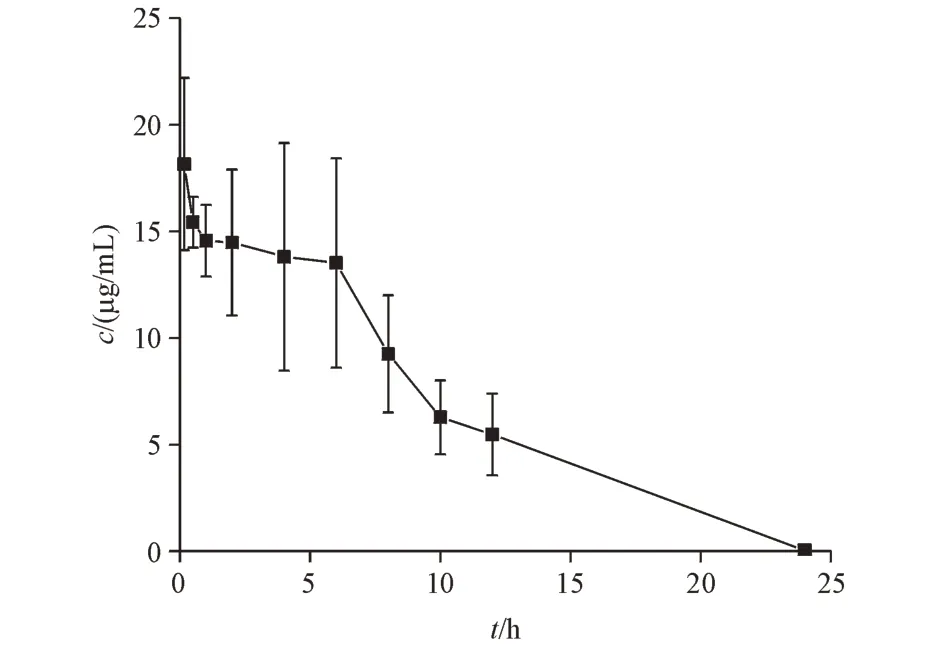
Figure 4 Profile of rifampicin in rat plasma concentration after iv ad‑ministration(± s,n = 6)
根据计算结果,一房室模型的R2为0.909 2,在所有拟合模型中最高,表明一房室模型为最优模型。最优模型拟合结果见图5。
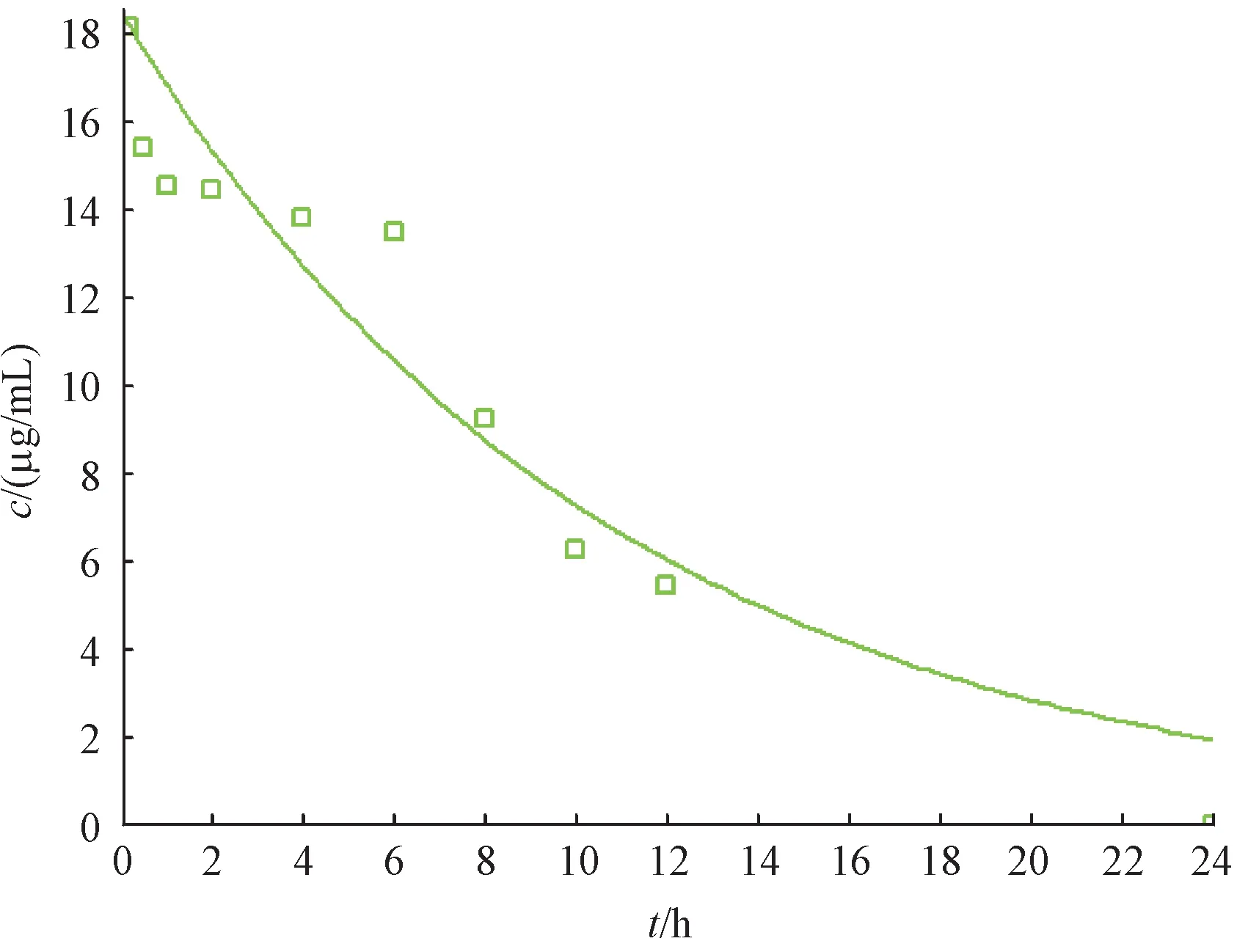
Figure 5 Atrioventricular model fitted by GastroPlusTM
虽然房室模型能够更好地体现出药物的药动学特征,但选择不同的房室模型会得到不同的药动学参数,进而对药物制剂等效性的判断产生影响,而非房室模型采用统计矩的方法,不依赖于药动学特征,更适合用于评价生物等效性。因此本研究选择非房室模型表征药物的药动学行为,计算得药动学参数:AUC0-t为169.6 μg·h/mL、MRT 为6.326 h、CL为0.029 L/h、Vss为0.186 h、t1/2为1.691 h。
3.2 建立并优化大鼠口服给药PBPK模型
3.2.1 口服给药药动学研究 选取健康的体重约为250 g雄性Wistar大鼠12只。实验期间饲养温度控制在(20 ± 5)℃、饲养湿度控制在30% ~ 70%。大鼠给药前禁食24 h,自由饮水。晶型Ⅰ与晶型Ⅱ利福平分别用1%CMC-Na 溶液配成2 mg/mL 的混悬液。将大鼠随机分为两组,按20 mg/kg 的给药剂量进行灌胃。
分别于大鼠给药后的0.16,0.5,1,2,4,6,8,10,12,24 h 用毛细玻璃管从眼眶静脉丛取血,同“3.1”项用高效液相色谱法进行血药浓度测定。最终测定的两种晶型利福平口服给药后血药浓度-时间曲线见图6,相关药动学参数见表2。
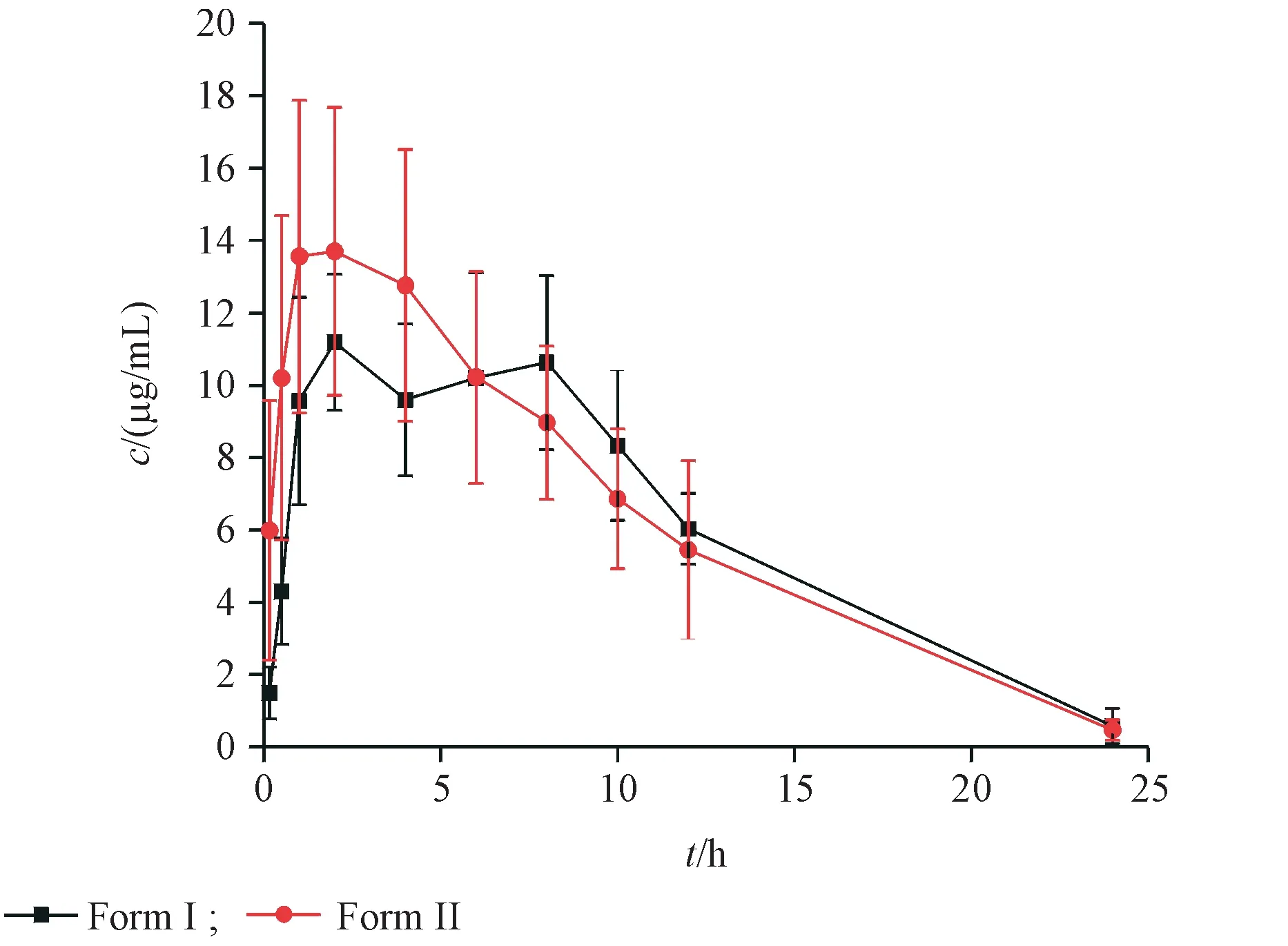
Figure 6 Profiles of rifampicin in rat plasma concentration after oral administration(± s,n = 6)
Table 2 Pharmacokinetic parameters of oral administration in rats(± s,n = 6)

Table 2 Pharmacokinetic parameters of oral administration in rats(± s,n = 6)
Parameter cmax/(μg/mL)tmax/h AUC0-t/(μg·h/mL)Form I 11.19±1.81 2.00±0.66 149.36±20.52 Form II 13.70±2.78 2.00±0.60 155.11±40.81
口服给药的利福平在被吸收后,主要通过肝脏经去乙酰化进行代谢。原型利福平与去乙酰化利福平会通过胆汁排泄进入肠道,在肠道再次被吸收入血,形成肝肠循环[13]。由于存在这种多次吸收入血的机制,会使血药浓度出现先下降然后再次上升的情况,表现在血药浓度-时间曲线上就是出现双峰甚至多峰。根据实验结果,Ⅰ晶型血药浓度-时间曲线中该现象较为明显。
通过计算大鼠口服两种晶型的利福平后的药动学参数,发现两种晶型的用于评价生物利用的关键药动学参数cmax,tmax与AUC0-t并无显著差异。
结合静脉给药的实验数据,根据绝对生物利用度公式:

式(1)中,AUC 为血药浓度-时间曲线下面积(μg·h/mL),下标po和iv 分别代表口服和静脉给药,D代表给药剂量(mg/kg)。计算得晶型I利福平的绝对生物利用度为88.82%,晶型Ⅱ利福平的绝对生物利用度为89.35%。由计算结果可知,虽然利福平是BCS Ⅱ类药物,但它的绝对生物利用度很高,两种晶型均可达到约90%。
3.2.2 建立并优化大鼠口服给药的PBPK 模型在大鼠静脉给药PBPK 模型的基础上,利用大鼠口服给药后的实测药动学数据来建立和优化大鼠口服PBPK模型。
根据静脉给药所得的药动学参数拟合口服数据,发现拟合的cmax比实测数据高,这是因为利福平具有肝脏首过效应,需要进行考虑。将肝脏首过效应设置为15%后,拟合结果良好。晶型Ⅰ与晶型Ⅱ口服给药的预测结果见图7。
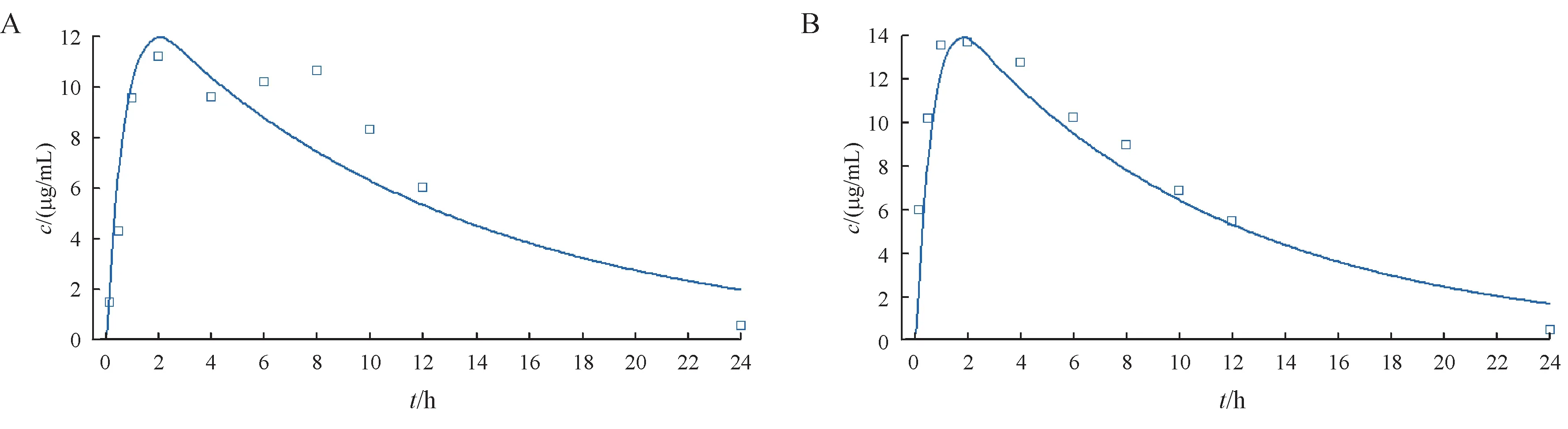
Figure 7 Stimulated and observed plasma concentration for two crystal forms of rifampicin in rat(po)
根据优化后的PBPK 模型,计算药动学参数,观测值与预测值对比结果见表3。结果显示,优化后的大鼠口服PBPK 模型的观测值与预测值非常接近,说明该模型构建合理。
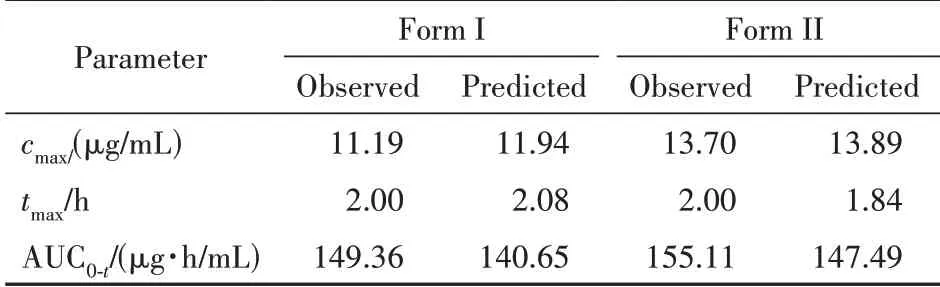
Table 3 Comparison of pharmacokinetic parameters of oral adminis‑tration in rats
利用该PBPK 模型预测两种晶型的利福平在大鼠体内不同部位的吸收情况,两种晶型之间的对比见表4。根据预测结果,利福平口服给药后在肠道吸收完全,且两种晶型吸收情况非常相似,都表现为在胃部不吸收,在十二指肠、空肠和盲肠分别吸收约18%、65%和15%,在回肠几乎不吸收。
3.3 大鼠口服给药PBPK模型种属外推
3.3.1 种属外推至健康人体 不同种属之间的清除率(CL)推算,主要采用异速增长公式,如式(2),通常情况下异速增长指数(b)的经验值为0.78[13]。
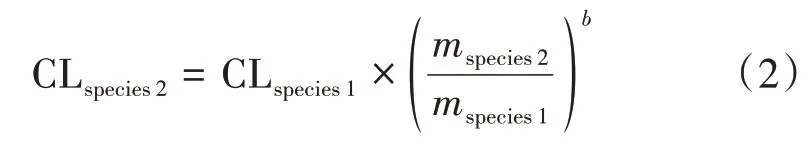
可利用式(2),通过已得的利福平在大鼠体内的血浆清除率,进而推算人体的血浆清除率。但是由于不同药物在不同物种内的处置过程差异很大,所以这种推算的方法通用但不准确,更推荐采用人体实测数据来得到清除率与分布容积。本研究选择利用在FDA 查询到的RIFADIN®IV(rifampin for injection USP)的审评文件[14]中报道的静脉滴注血药浓度数据(具体血药浓度数据见表5)来获取人体的清除率和分布容积,再通过GastroPlusTM软件,利用优化好的大鼠口服PBPK 模型来种属外推至健康人体,得到健康人体的口服PBPK模型。
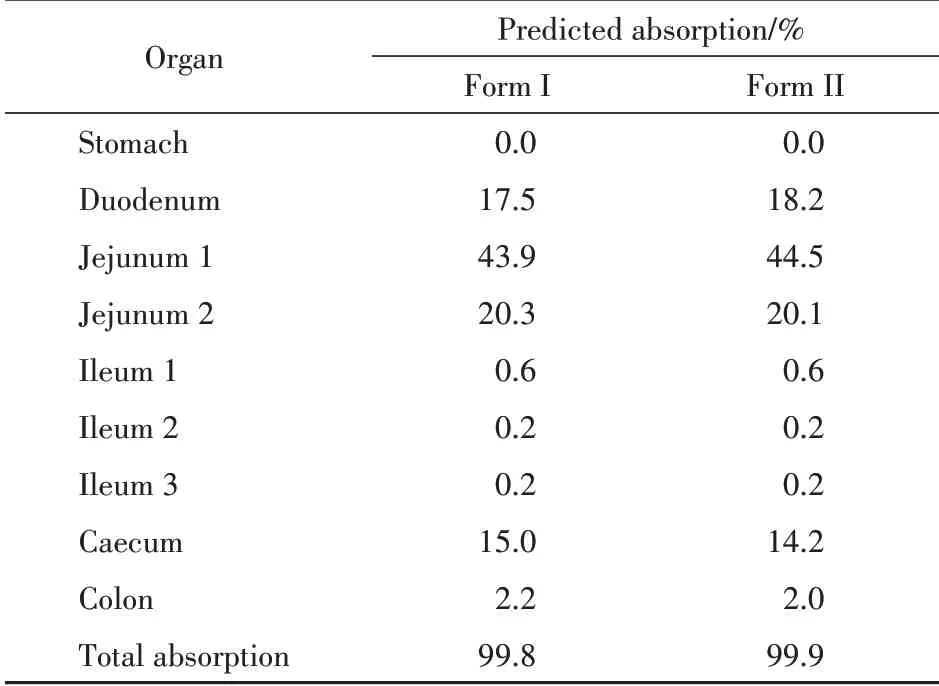
Table 4 Amount predicted of drug absorption in various intestinal segments after oral administration in rat
Table 5 Plasma concentrations of healthy people by intravenous infu‑sion of 600 mg(infusion time 30 min,± s)
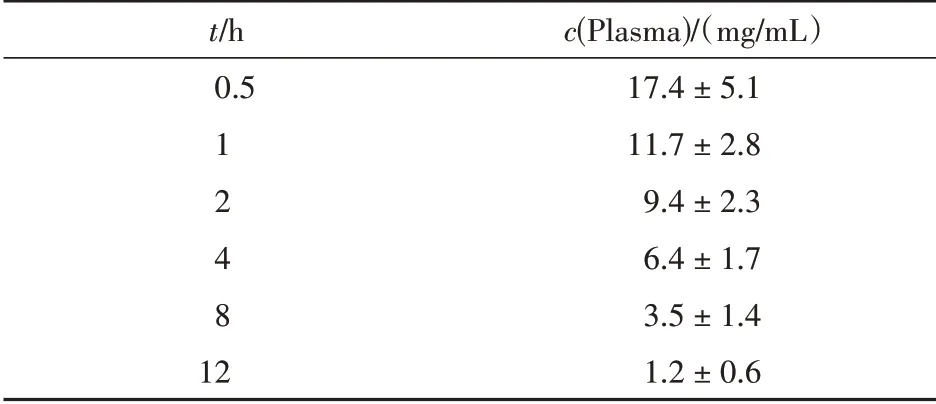
Table 5 Plasma concentrations of healthy people by intravenous infu‑sion of 600 mg(infusion time 30 min,± s)
t/h 0.5 1 2 4 8 12 c(Plasma)/(mg/mL)17.4±5.1 11.7±2.8 9.4±2.3 6.4±1.7 3.5±1.4 1.2±0.6
根据FDA 审评文件提供的数据,计算得药动学参数:AUC0-t为67.18 μg·h/mL、MRT 为4.348 h、CL为0.14 L·kg/h、Vss为0.607 L/kg、t1/2为2.590 h。
3.4 基于健康人体口服给药PBPK模型的预测
3.4.1 构建生理模型及设计给药方案 选择体重为70 kg 的健康男性,利福平给药剂型选择目前较为常见的胶囊,给药剂量定为600 mg,假设给药时需服用水200 mL。血浆蛋白结合率定为80%[15],血浆占全血百分比取平均值55%。
3.4.2 预测结果 利用种属外推得到的人体口服PBPK 模型,预测健康人体口服不同晶型的利福平胶囊后的血药浓度-时间曲线、药动学参数及吸收情况。
口服不同晶型的利福平胶囊600 mg 的模型预测结果:药动学参数见表6,血药浓度-时间曲线见图8,吸收情况见表7。
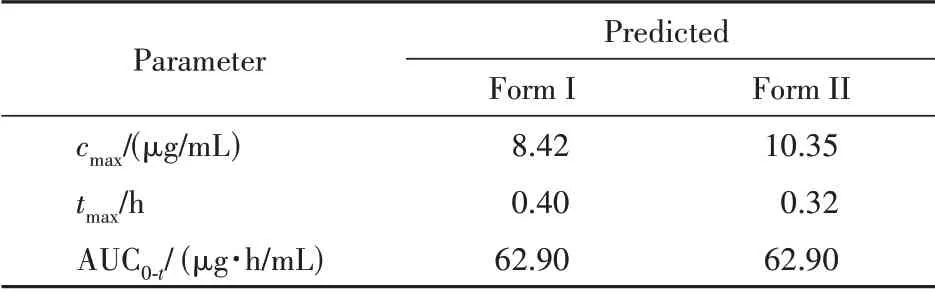
Table 6 Comparison of pharmacokinetic parameters of oral adminis‑tration in healthy people
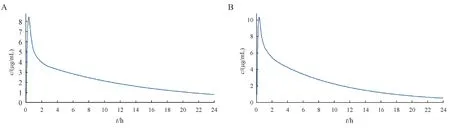
Figure 8 Stimulated plasma concentration for two crystal forms of rifampicin in healthy people (po)
根据本研究建立的PBPK 模型对两种晶型的利福平吸收部位的预测,两种晶型的利福平在各个肠段的吸收情况并无显著差异。根据预测结果,晶型Ⅰ与晶型Ⅱ两种利福平在人体的血药浓度-时间曲线、药动学参数和吸收情况非常相似,推断两种晶型的利福平具有生物等效性。
4 讨 论
本研究先对两种晶型的利福平进行了体外研究,结果表明晶型Ⅰ与晶型Ⅱ利福平虽然溶解特征相似,都表现出明显的pH 依赖性,但两者除了在溶解度较高的pH 1.0介质中的溶出行为不存在差异外,在其他3 种介质中,晶型Ⅰ的溶出速度都显著慢于晶型Ⅱ。由于两种晶型的利福平的溶出曲线并不相似,所以不能通过体外溶出实验来进行生物利用度豁免,在制剂评价时需要进行生物等效性实验。
在此体外研究的基础上,本研究采用了搭建PBPK 模型的方法来进行体内预测。根据PBPK 模型对两种晶型利福平的吸收部位及吸收量进行预测,发现无论是大鼠还是健康人体,两种晶型的利福平在酸性环境的胃部都没有吸收,但在肠道能够被完全吸收。利福平作为一种BCS Ⅱ类药物,根据其低溶解性和高渗透性的特点判断出影响其生物利用度的关键步骤是制剂的溶出,但由于利福平表现出的溶解特点与吸收特点,无论何种晶型的利福平在强酸性的胃部环境中都能快速溶解完全,然后由于其高渗透性在肠道被迅速吸收。正是胃内溶解完全和肠道吸收迅速的两个特点,使得两种晶型的利福平在pH 4.0、pH 6.8 和水这3 种介质中的溶出行为不一致的情况下,也会由于其在pH 1.0 介质中迅速地溶出,而表现出体内药动学行为的相似和生物利用度的等效。

Table 7 Amount predicted of drug absorption in various intestinal segments after oral administration in human
在仿制药研发过程中,生物等效性实验作为最终准则,存在周期长、费用高、风险大等问题,因此,如何在实验进行之前规避风险就变得至关重要。本研究提供了一种新思路:在进行体外溶出评价的基础上,建立PBPK 模型进行体内预测,仅进行少量动物试验就能够达到预测人体生物等效性的目的,有效降低生物等效性实验失败的风险。模型预测的方法并不能完全代替人体实验,模型模拟的准确度需要用准确可靠数据加以支持,但随着对PBPK 模型的深入研究,这一降低生物等效性实验风险的建模策略将会得到更广泛的应用。
猜你喜欢
临床肝胆病杂志(2022年2期)2022-11-24
中南药学(2022年2期)2022-11-14
四川生理科学杂志(2022年9期)2022-10-10
中国现代医生(2022年6期)2022-04-23
中国药学药品知识仓库(2022年5期)2022-04-11
心理与健康(2020年3期)2020-03-25
中国中药杂志(2016年23期)2017-04-07
医学信息(2016年32期)2017-02-22
中国中药杂志(2016年22期)2017-02-13
上海医药(2016年1期)2016-02-22
