
靶向MLL1-WDR5蛋白-蛋白相互作用抑制剂的研究进展
2022-05-09 02:58尤启冬郭小可
中国药科大学学报 2022年2期
陈 鑫,杨 倩,尤启冬,郭小可*
(1中国药科大学江苏省药物分子设计与成药性优化重点实验室,南京 211198;2中国药科大学药学院,南京 211198)
表观遗传学是近年来生命科学界最热门的领域之一,其涉及的生物学活动覆盖了从胚胎发育到动物毛发颜色调节等各个环节。表观遗传还参与众多疾病的起始、发展和维持,如:糖尿病[1]、精神疾病[2]、肿瘤[3]等。通过干涉表观遗传调节过程来调控疾病的发展受到药物化学家们的关注[4-5]。表观遗传指的是DNA 碱基序列不变的可遗传性状变化,包括DNA 修饰、组蛋白修饰、非编码RNA 修饰、染色质重组和核小体定位等[6-7]。其中,组蛋白修饰包括甲基化、乙酰化、磷酸化和泛素化等。在众多组蛋白修饰中,组蛋白甲基化是研究最为深入的修饰之一[8]。由于组蛋白甲基化异常,导致肿瘤相关的基因转录异常,是诱发肿瘤的重要因素[9]。因此选择性靶向组蛋白甲基化过程、逆转基因的异常表达,成为治疗肿瘤的新策略之一。
SET1 家族是负责调节组蛋白H3 的第4 位赖氨酸(H3K4)甲基化过程的蛋白家族,因其特有的SET 结构域而得名。SET1 家族可以通过将甲基载体S-5'-腺苷-L-蛋氨酸(SAM)上的甲基转移至H3K4 而实现组蛋白的甲基化。SET1 家族成员有MLL1-4(mixed lineage leukemia 1-4)、SET1a 和SET1b,其中MLL1 是研究最为深入的一个成员。研究发现,组蛋白甲基转移酶MLL1 与多种肿瘤的发生、发展相关,成为近年来治疗肿瘤的热门靶标[10-13]。目前,已有多种靶向MLL1 甲基转移酶复合物的抑制剂被研究和开发,其中部分抑制剂已推进至临床研究阶段,如DOT1L(disruptor of telomeric silencing 1-like)抑 制 剂EPZ-5676[14]和MENIN(multiple endocrine neoplasia type 1)抑制剂SNDX-5613[15]。MLL1 和WDR5(WD40 repeat 5)的蛋白-蛋白相互作用(protein-protein interaction,PPI)是MLL1 甲基转移酶发挥正常生理功能的基础,近些年来成为药物分子研究的热点。本文主要针对MLL1-WDR5 PPI在肿瘤中的重要作用以及其抑制剂的进展进行系统总结,期望能为该领域研究人员提供参考。
1 MLL1-WDR5的生理功能
1.1 野生型MLL1(WT-MLL1)
MLL1 又称组蛋白赖氨酸甲基转移酶2(KMT2A),因其可诱发混合谱系白血病(mixed lineage leukemia)得名[16]。MLL1 在人体组织中广泛存在,编码MLL1 的基因位于11q23 染色体上。在MLL1基因被翻译后,全长MLL1蛋白在第2 666/2 667 残基和第2 718/2 719 处被taspase1 切断,产生两个多肽(N 端多肽和C 端多肽,MLL-C 和MLLN),随后N 端多肽和C 端多肽分别通过FYRN 和FYRC 连接形成一个完整的活性MLL1 蛋白(图1)[17-22]。
MLL1 的主要生物学功能是在胚胎发育、造血和神经发育过程中通过调节组蛋白H3K4 的甲基化状态,维持下游Hox(homeobox)基因表达,而Hox基因在造血功能、干细胞自我更新等生理过程中起着重要作用[23-24]。MLL1还能识别二甲基化和三甲 基 化 的H3K4(H3K4me2 和H3K4me3),招 募MLL1 到靶基因,进一步激活下游Hox基因的表达[25-26]。在婴儿发育期间,MLL促进造血干细胞的分化,在成人体内,MLL 帮助维持血细胞更新[27]。此外,MLL1 还参与了血管的生成[28]、调控肿瘤抑制因子以及参与组织生长和发育相关基因的调节[29]。
1.2 MLL1融合蛋白
混合谱系白血病基因(MLLgene)是三胸腺果蝇基因(drosophila trithorax)的同源基因[30]。MLL基因可与其他基因重排,这种现象在白血病患者中广泛存在,约10%的白血病患体内存在MLL基因重排现象[31]。MLL基因重排与急性髓性白血病(AML)、淋巴性白血病(ALL)、双表型或混合性白血病的发生密切相关。MLL基因重排主要发生在1 岁以下婴儿(主要为ALL)和中青年患者(主要为AML)中[31]。MLL基因重排有两种类型:MLL基因特异性突变,包括基因内部缺失、部分序列重复串联(partial tandem duplications,MLL-PTD)或MLL全基因重复[16,24,32];第二种是由于染色体易位导致11q23 染色体(MLL基因)和其他染色体重组,产生MLL 融合基因(MLL-FP)[19]。MLL重排主要发生在MLL基因的RD 和PHD 结构域之间。MLL基因可与80 多个不同的伴侣基因融合,融合的伴侣基因大多数都编码转录因子,诱发白血病的MLL 融合蛋白主要来源于6 个融合基因:MLL-AF4、MLL-AF6、MLL-AF9、MLL-AF10、MLL-ELL和MLLENL[33]。MLL 融合蛋白缺乏C 端TAD、Win 和SET结构域,因此失去了WT-MLL1-C端的相关功能,缺乏SET 结构导致MLL-FPs 无法催化H3K4 甲基化;在WT-MLL1-N 结构中保留AT-钩(AT-hook)结构域、CXXC 域、MBD 和SNL,使其依然保留了识别和结合相关组蛋白甲基化标记的能力(图1 和图2)。然而,由于其他伴侣基因的融合,MLL-FPs 从伴侣基因中获得新的功能[19]。虽然MLL-FPs 诱发肿瘤的机制尚未完全确定,但已有研究表明,部分MLLFPs可以通过调节RNA聚合酶Ⅱ,诱导相关基因异常表达,继而诱发白血病[34];MLL-FPs 还通过招募其他蛋白特异性激活下游基因表达,导致下游基因表达异常,进而诱导或促进白血病进程[31,35-36]。总之,MLL-FPs可以通过多种机制导致白血病的发生,维持白血病的发展。

图1 MLL1蛋白翻译后修饰和MLL1甲基转移酶复合物

图2 染色体重组导致MLL1融合蛋白的形成
1.3 MLL1-WDR5 PPI
WT-MLL1是MLL1-FPs相关白血病发生、发展和维持所不可或缺的。MLL基因重排仅发生在一个等位基因上,MLL1-FPs的靶基因是WT-MLL1靶基因的子集。若野生型MLL基因被敲除,仅MLL1-FPs 无法诱导和维持白血病[37-39]。首先,MLL1-FPs缺少了WT-MLL1 的C 端部分,缺乏与转录调控相关结构域,如PHD3结构域(图2)。PHD3可识别并结 合H3K4me2 和H3K4me3。WT-MLL1 的PHD3通过与CXXC-PAFc 协同工作,招募野生型MLL1复合物到Hox基因,激活Hox基因,并阻止调控基因表达的遏制剂“ESET”和Hox基因结合,创造一个更适合MLL1-FPs 结合的染色质状态。在WTMLL1 存在的情况下,MLL-AF9 等MLL1-FPs 被招募到Hox基因,持续激活Hox基因进而诱导白血病发 生[25]。H3K4me2 和H3K4me3 标 记 需 要WTMLL1 复合物维持完整的生理功能。H3K4me2 和H3K4me3 标记是MLL1-FPs 的主要结合位点,降低染色质H3K4me2 和H3K4me3 水平可以影响MLL1-FPs对肿瘤的诱导作用[38-39]。
破坏MLL1 甲基转移酶复合物正常的生理功能,降低H3K4 的甲基化水平,减少MLL1-FPs 的结合位点,从而降低MLL1-FPs 被招募到靶基因的概率是治疗融合蛋白诱导的白血病的策略之一。WDR5 是MLL1 甲基转移酶复合物的核心组件,是MLL1 复合物发挥正常生理功能的基石之一。WDR5 通过和MLL1 的Win 序列相互作用,招募MLL1 复合物的其他成员(RbBP5、Ash2L 和DPY-30)组成功能完整的MLL1 甲基转移酶复合物(图1-B),并稳定其构象[40-41]。单独的MLL1 蛋白只能缓慢地催化H3K4 的单甲基化。在缺乏WDR5 的情况下,即使其他MLL1 复合物组成部分存在,MLL1 也无法发挥其完整的甲基转移酶活性[40-41]。因此WDR5 是MLL1 发挥完整的甲基转移酶活性以及进一步二甲基化和三甲基化H3K4的必要条件。阻断MLL1-WDR5 PPI,降低MLL1甲基转移酶活性,进一步降低组蛋白H3K4 的甲基化是治疗MLL1-FPs 诱导白血病的潜在方法。而近些年小分子抑制剂的研发也证明了该方法的可行性[42]。
2 靶向MLL1-WDR5 PPI的抑制剂
2.1 MLL1-WDR5 PPI抑制剂的作用位点
WDR5 是典型的WD40 重复域蛋白家族的成员。总体呈游泳圈状,有7 个反式β 螺旋桨域,每个叶片由4 个反式平行的β 折叠组成,其中每个叶片以保守的丝氨酸-组氨酸(SH)开始,以色氨酸-天冬氨酸(WD)结束,大小为34 kD。7个结构域环绕在一起组成一个贯穿蛋白质顶部和底部的中心腔(图3)[43-44]。

图3 WDR5的结构和MLL1-WDR5 PPI抑制剂的主要作用位点
MLL1-WDR5相互作用界面位于WDR5蛋白的顶部,被7 个七叶β-螺旋桨域包围[45-46]。WDR5 的该空腔和MLL1的Win序列(MLL1上一段包含精氨酸的保守序列)相互作用,故又被成为Win-Site,此位点是干扰MLL1-WDR5 蛋白-蛋白相互作用抑制剂的主要作用位点。本文中根据小分子与WDR5蛋白的结合方式将此位点的结合界面分为5个结合口袋——P1~P5,其中,P1是位于空腔深处的精氨酸结合口袋,又称为氢键网络区。该口袋是特征口袋,所有MLL1-WDR5 PPI 抑制剂结构中都包含了能和P1口袋相互作用的碱性基团或精氨酸模拟片段[47-48]。P2是一个疏水口袋,而P3与P2相邻,P3中的极性氨基酸残基Asp107对小分子与蛋白的结合有巨大的贡献。P4 由Phe133、Phe149 和Asp107 组成,P5 由Tyr191、Cys261、Tyr260 和Lys259 组成的疏水口袋。P5是小分子与WDR5相互作用的热区。整个相互作用界面是可塑的,小分子和蛋白的结合对口袋的形状变化都有诱导作用[47,49]。
靶向MLL1-WDR5 PPI 抑制剂最早报道于2010 年,距今已有十多年的研发历史。根据小分子的结构和类型分类主要有拟肽类、多取代苯基哌嗪类、芳香五元杂环类(后两者的分类主要依据氢键网络区的结合基团结构而定)以及基于这些化合物衍生的工具分子(图4)。

图4 不同类型的代表MLL1-WDR5蛋白-蛋白相互作用(PPI)抑制剂
2.2 拟肽类MLL1-WDR5 PPI抑制剂
肽类和拟肽类小分子抑制剂主要由美国密歇根大学的王少萌课题组开发。其通过使用多肽的基序简化,将MLL1 中的WIN motif 简化为三肽Ac-ARA-NH2(Ki=120 nmol/L)[50-51]。为进一步提高三肽活性,王少萌课题组替换并修饰了Ala1 和Ala3,然后在N-端和C-端引入合适的疏水基团,得到拟肽MM-101、MM-102 和MM-103(图5)。MM-101 和MM-102 的精氨酸胍基深入氢键结合口袋P1,疏水碳链和脂肪环占据了P2、P3两个疏水口袋,其活性提高了200倍(IC50=2.4 nmol/L)。MM-102对MLL1 H3K4 甲基转移酶(MLL1 HMT)的抑制活性并不高(IC50=141 μmol/L),但是它仍能降低MLL1介导的白血病发生过程中重要基因HoxA9和Meis-1的表达,并且可以选择性抑制MLL1 融合型白血病细胞的生长以及诱导细胞凋亡,而对只含WT-MLL1 蛋白的白血病细胞作用较弱。MM-102 首次证实了靶向MLL1-WDR5 PPI的可行性和有效性[42]。
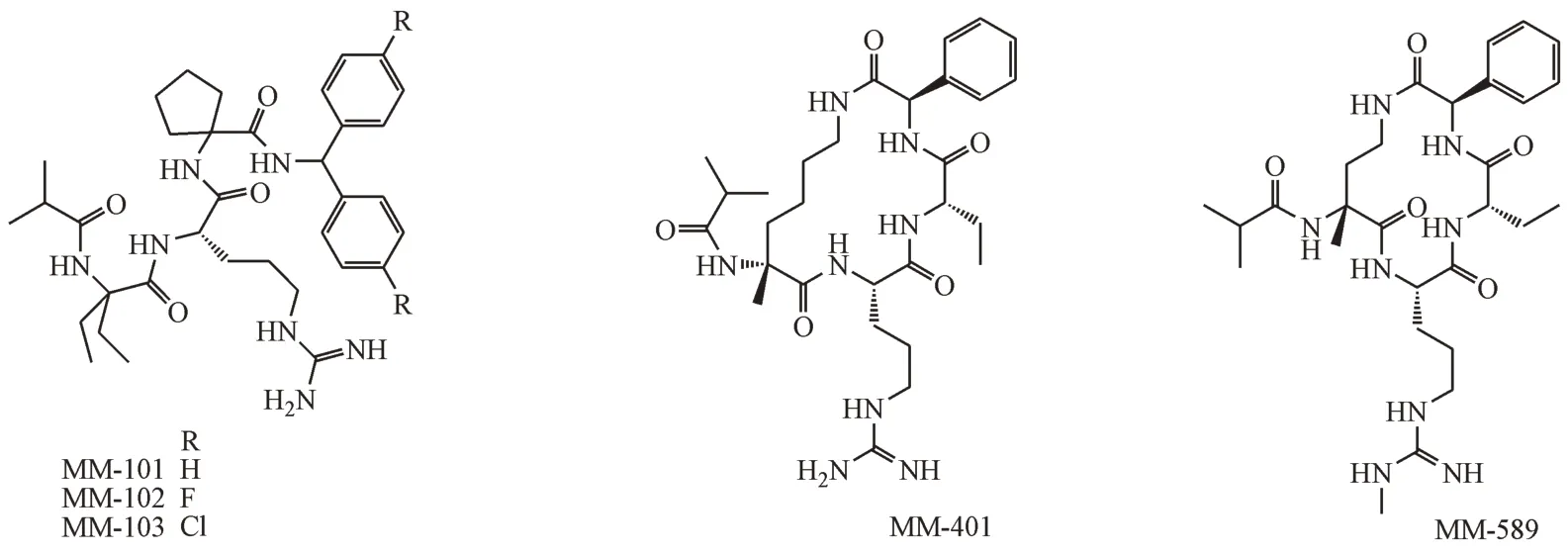
图5 拟肽类MLL1-WDR5 PPI抑制剂
王少萌课题组继续对拟肽MM-101 进行改造得到了环状拟肽MM-401(IC50= 0.9 nmol/L,图5)。将MM-401 连接成环的脂肪链代替了MM-102 疏水链的作用,和其他疏水侧链共同占据了P4 口袋(图6-A),MM-401 的甲基转移酶活性(IC50= 0.32 μmol/L)相比于MM-101 和MM-102 提高近400倍。MM-401对含有MLL融合基因的人白血 病 细 胞 株(GI50MV4-11(MLL-AF4)=12.42 μmol/L,GI50Molm13(MLL-AF9)= 23.97 μmol/L,GI50Kopn8(MLL-enl)=29.73 μmol/L)的抑制能力有大幅度的提高,但是依然处于中等水平[52]。基于MM-401,他们进一步在精氨酸胍基末端N 原子引入甲基,并缩短环肽连接链得到MM-589。甲基取代的胍基通过水介导的氢键和P1 相互作用。MM-589 以更小的环形成一个更紧凑的立体结构,几个分子内氢键降低构象的灵活性,使苯环以近乎平行的方式和Tyr260 的苯环相互作用(图6-B)。MM-589 靶标活性没有显著的提高,但是其MLL1 甲基转移酶活性大幅提高,IC50为12.7 nmol/L,其代谢稳定性适中(t1/2= 60 min)。MM-589 对具有MLL易位的人AML 细胞系Molm13 和MV4-11 细胞的抑制IC50分别达到0.21 和0.25 μmol/L,有良好的选择性,是迄今为止报道的活性最高的靶向MLL1-WDR5 PPI的拟肽抑制剂[53]。

图6 WDR5与多肽和拟态的结合模式对比
2.3 多取代苯基哌嗪类MLL1-WDR5 PPI抑制剂
多取代苯基哌嗪类化合物来自于筛选得到的小分子化合物WDR5-0102,该化合物由Guillermo课题组[54]基于荧光偏振高通量筛选所得,经过Al-Awar 课题组[55]进一步修饰,得到了甲基哌嗪苯类的先导化合物WDR5-47(图7)。多取代苯基哌嗪类化合物根据苯环上取代基的不同又可以细分为苯甲酰胺类、嘧啶氨基苯类和三氟甲基吡啶酮类。

图7 苯甲酰胺类MLL1-WDR5 PPI抑制剂
2.3.1 苯甲酰胺类化合物 WDR5-0102 结构中包含了和精氨酸结合口袋结合的N-甲基哌嗪,哌嗪和中心苯环相连,邻位则是苯甲酰氨基。因此WDR5-0102是第一个苯甲酰胺类化合物。WDR5-0102 靶标结合活性适中、结构简单,是优良的苗头化合物(Kd= 3.0 ± 1 μmol/L)[54]。在WDR5-0102的基础上,Al-Awar 课题组通过晶体结构指导结构改造,他们保留了WDR5-0102 上的N-甲基哌嗪,进一步修饰了苯甲酰胺部分的2-氯苯基,原苯环3 位和4 位分别引入甲基和F 原子,得到了WDR5-47。WDR5-47 的靶标结合活性提高25 倍(IC50=0.27±0.1 μmol/L)。其甲基和F 原子占据了P2 和P3,增强了口袋中疏水相互作用。在生理pH 条件下,质子化的N-甲基哌嗪基锚定在P1 底部,酰胺键与Ser91的侧链和Cys261的主链氮原子分别形成直接和间接的氢键相互作用(图8)[55]。
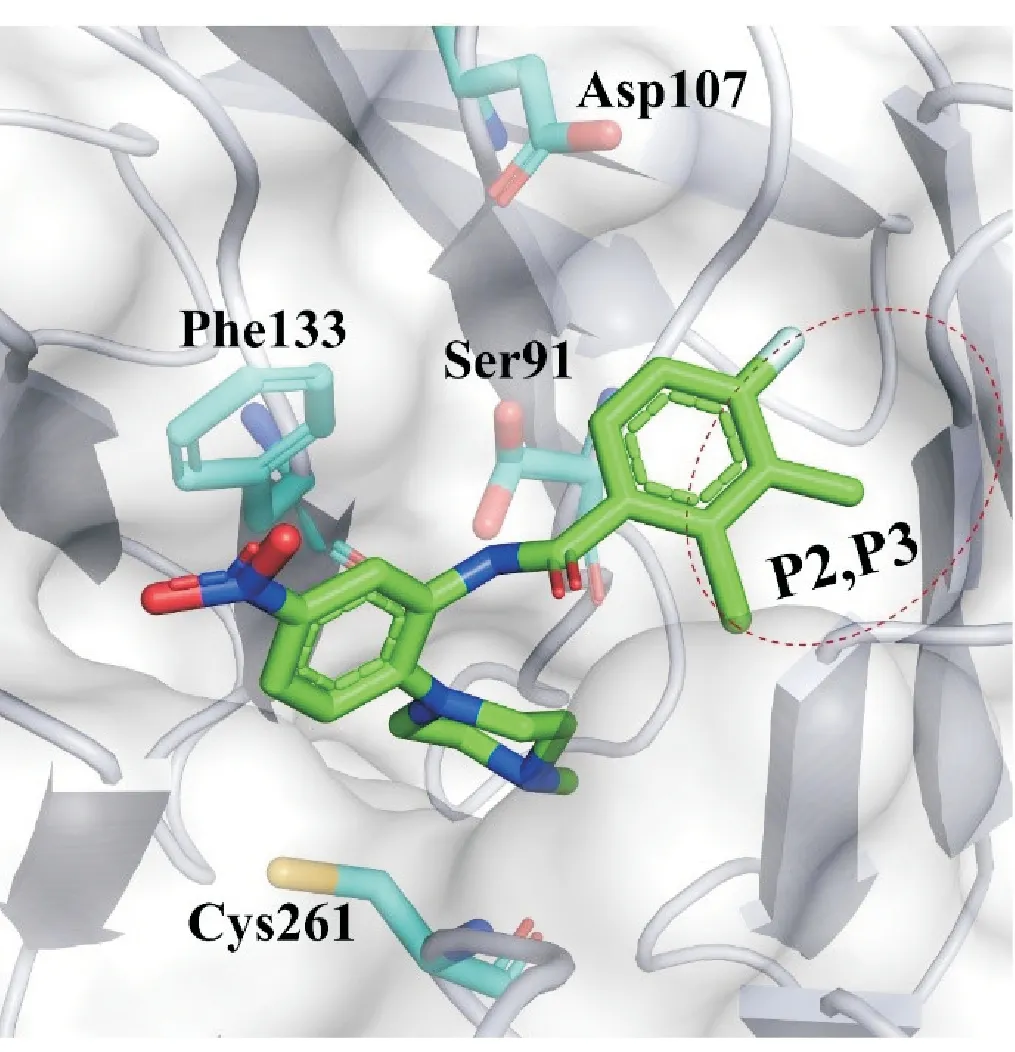
图8 WDR5-47和WDR5的结合模式(PBD:4IA9)
基于WDR5-47,中国药科大学的尤启冬课题组在WDR5-47 的中心苯环的5 位使用吡啶替换硝基得到了化合物23,其靶标活性进一步提升(IC50=104 nmol/L)[56]。之后尤启冬课题组又将吡啶替换为4-氨酰苯基,并在原苯酰胺苯环5′位上引入氨基得到DDO-2117(图7)。DDO-2117 苯酰胺上的氨基通过氢键与Asp107相互作用。长链的伯胺深入溶剂区,增强和溶剂区的作用。DDO-2117 具有较好的靶标活性(Kd= 13.6 nmol/L,IC50= 7.6 nmol/L),可以显著抑制MV4-11 细胞的生长(GI50=7.6 μmol/L)[56]。
除DDO-2117 外,尤启冬课题组还使用其他策略对WDR5-47 进行优化,获得了2 个不同结构的苯甲酰类化合物(图7):DDO-2093 和WL-15。DDO-2093 和WL-15 分别使用三氮唑和酚酯基取代DDO-2117 的中心苯环上的氨酰苯基,DDO-2093 有纳摩尔范围的靶标活性(IC50<10 nmol/L),并且有中等的细胞抗增殖活性。这两类化合物都有较好的甲基转移酶抑制活性和体外抗增殖活性,都可以下调Hox和Meis-1基因,并诱导白血病细胞凋亡。其中,DDO-2093 相比于DDO-2117,其水溶性和安全性均得到较大提高,其首次在小鼠体内移植瘤模型中证明了有效性[57]。
2.3.2 嘧啶氨基苯类化合物 同样基于化合物WDR5-47,尤启冬课题组使用骨架跃迁策略得到了嘧啶氨基苯类化合物,通过将WDR5-47 的苯甲酰胺基团替换为4-氯-5-氨基取代嘧啶,中心苯环硝基取代为4-(N,N-二甲氨酰)苯基,并在中心苯环5 号位引入F 原子得到了DDO-2213(图9)。DDO-2213 有较好的靶标活性,和DDO-2093 一样,DDO-2213 具有中等的MLL1 甲基转移酶抑制活性以及体外抗增殖活性,可以下调Hox和Meis-1基因,并诱导白血病细胞凋亡。同时,DDO-2213 的有效性也在小鼠体内移植瘤模型中得到证明[58]。
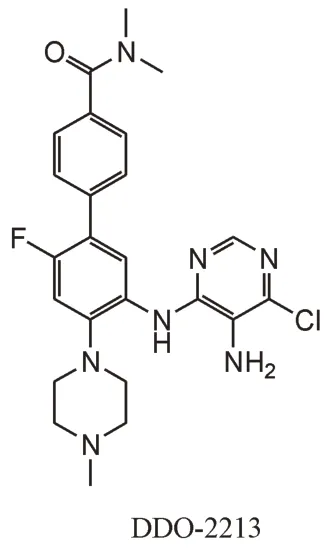
图9 嘧啶氨基苯类MLL1-WDR5 PPI抑制剂
2.3.3 三氟甲基吡啶酮类化合物 在WDR5-47的结构基础上,Al-Awar 课题组根据片段筛选,将取代苯甲酰胺替换为4-(三氟甲基)吡啶酮,并将WDR5-47 的硝基替换为3-甲基吗啉取代的苯环得到OICR-9429(图10),这是第1 个三氟甲基吡啶酮类化合物。与WDR5-47 相比,OICR-9429 有更高的靶标活性(Kd= 0.03 μmol/L)、更优的溶解度和更佳的选择性。此外,OICR-9429 不仅能够抑制MLL1 融合蛋白型白血病的细胞株的生长,还可抑制表达带有N 末端C/EBPα 突变(IC50≈5 μmol/L)的p30的原代人AML细胞的增殖[48]。
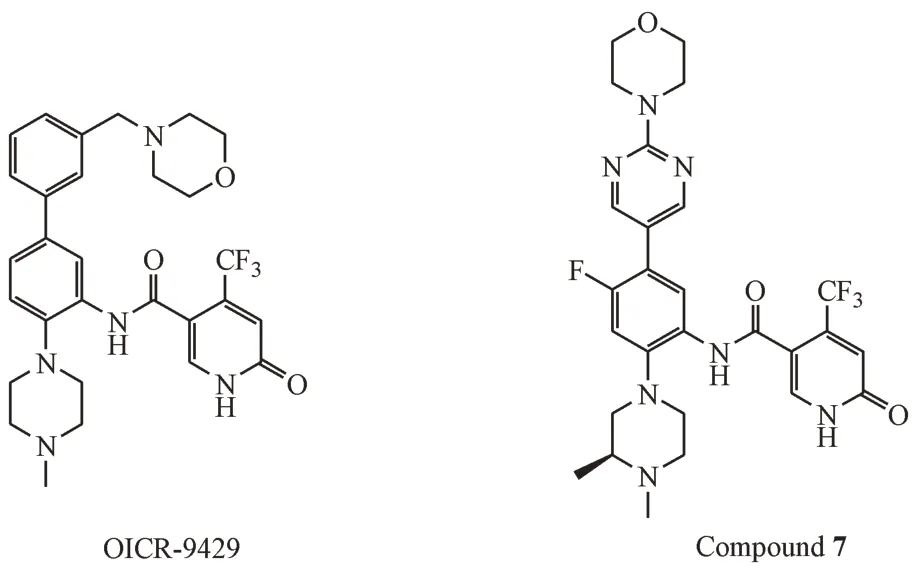
图10 三氟甲基吡啶酮类MLL1-WDR5 PPI抑制剂
在OICR-9429 的基础上,Al-Awar 课题组继续优化了靶标活性和成药性。在中心苯环C-4 处的引入F 原子,并使用4-(嘧啶-2-基)吗啉取代中心苯环C-5 处的4-苄基吗啉,同时在N-甲基哌嗪的2 号位引入甲基后得到化合物7。化合物7 是第一个皮摩尔级别的MLL1-WDR5 PPI 抑制剂(Kd=0.06 nmol/L),有小于百纳摩尔的体外抗增殖活性(MV4-11)[59]。关于化合物7 后期结构优化已有专利[60]报道,但在结构上改动较小且活性增强有限。
2.4 芳香五元杂环类MLL1-WDR5 PPI抑制剂
2018 年,Fesik 课题组[47,62]基于碎片筛选和碎片生长得到了新的芳香五元杂环类抑制剂。该类抑制剂的特征就是使用五元杂环替代以往抑制剂中的N-甲基哌嗪和精氨酸胍基与P1 口袋结合。相比于N-甲基哌嗪芳香五元杂环和P1 结合的更为深入,并诱导之前并未参与小分子相互作用的Phe263 和五元芳香杂环形成了π-π相互作用。相比于甲基哌嗪苯类化合物,芳香五元杂环类化合物分子整体柔性更强,且结合模式有较大的不同。
Fesik 课题组基于NMR 的片段筛选得到了抑制剂的基本片段——2-苯基-6,7-二氢-5H-吡咯并[1,2-a]咪唑。环状咪唑基团占据P1 并与Phe133和Phe263 形成π-π堆叠,咪唑3 号氮原子通过氢键和Cys261 的羰基相互作用。进而在苯环3 号位引入苄基取代的苯甲酰胺将化合物的结合区扩展P5,并通过构象限制进一步优化获得化合物6e(图11)。与OIRC-9429,DDO-2117 相比,化合物6e 和WDR5 的(PDB ID:6DAS)结合模式不同。化合物6e 是一类新的MLL1-WDR5 PPI 抑制剂,有较好的靶标活性(FPAKi<1 nmol/L,TR-FRETKi= 0.902 nmol/L),和中等的体外抗增殖活性(GI50(MV-4-11)=7.25 μmol/L)[47]。

图11 芳香五元杂环类MLL1-WDR5 PPI抑制剂
2019 年,Fesik 课题组基于碎片筛选得到了另一种结构的MLL1-WDR5 PPI 抑制剂。他们得到了初始精氨酸模拟片段——咪唑胺结构,使用苄基酰胺将分子延伸至P5,通过修饰苄基以增强和P5 的相互作用,并在中心苯环上引入F 指向P2 得到化合物C3。同时筛选到的另一个片段——5-(1H-咪唑-3-甲基)糠醛酸,他们将酸和苄胺缩合以利用苄基占据P5 口袋,并用2-氟-6-甲基吡啶代替F 来占据P2,在原有咪唑的C-2位引入亚氨基增强和氢键网络区的相互作用得到化合物C6,因此靶标活性也更高[61]。基于化合物C6,Fesik 课题组继续优化了和P2 和P5 结合的基团,替换原来的基团为4-氟-2-甲基苯和3,5-二甲氧基苯。然后中心苯环替换为二氢异喹啉酮,通过构象限制调整取代苄基和P5 的结合角和方向,并改善化合物的体内代谢稳定性,最终得到化合物16。晶体结构表明,咪唑亚胺深入P1口袋中,通过π-π堆叠相互作用被Phe133 和Phe263 夹在中间。亚胺氮原子通过与水分子和Cys261 形成双齿氢键,4-氟-2-甲基苯基和3,5-二甲氧基苯基分别占据的P2 和P5 口袋。化合物16和C6有相同水平的靶标活性,可诱导P53 表达和P53 依赖性细胞凋亡(图12)。相比化合物C6,化合物16 的细胞效价和成药性大幅提升,体外抗增殖活性GI50均小于100 nmol/L。除MLL 细胞系外,化合物16 还可抑制其他类型的肿瘤细胞的增殖,如胰腺导管癌细胞[62]。
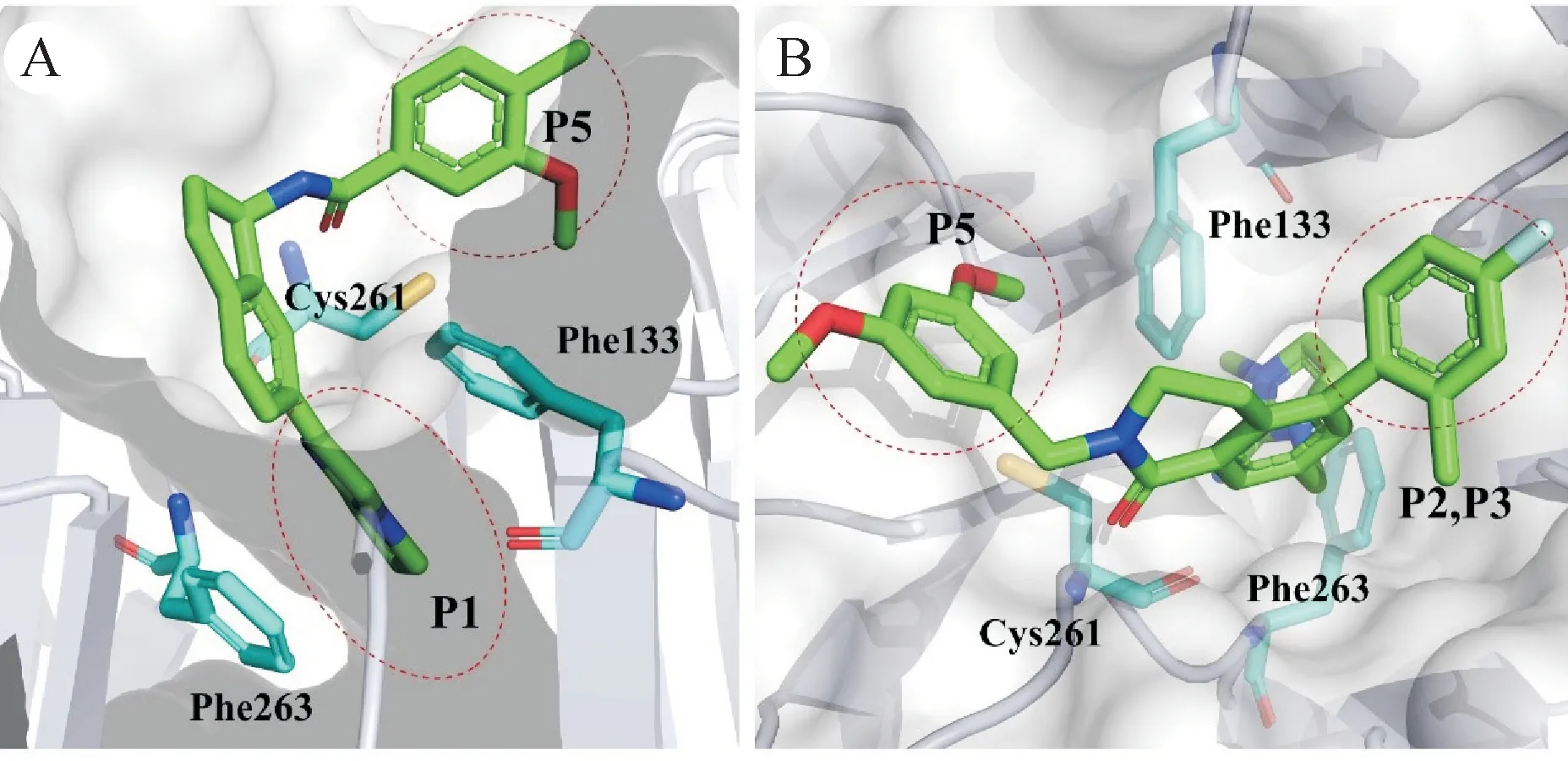
图12 WDR5和芳香五元杂环类抑制剂的相互作用
2.5 基于抑制剂的工具分子
2017 年,Vázquez 课题组报道了基于多肽类光调控WIN site 抑制剂。抑制剂基于WIN motif,引入4-(4′-氨甲基苯基偶氮)苯甲酸(AMPB)作为光调控元件,获得靶标活性较好的光调控制剂(Ki=1.24 nmol/L)。用光处理后,抑制剂的顺反异构发生改变,导致靶标活性变化,体外MLL1 的甲基转移酶活性也随之改变。化合物以微摩尔GI50抑制小鼠骨髓细胞的增殖,顺式和反式异构体之间有明显的活性差异[63]。2018 年,基于DDO-2117,使用连接链在末端游离氨基上引入生物素标签获得了小分子探针LC001。该探针靶标活性和DDO-2117 相近,IC50为17 nmol/L,具有类似于小分子抑制剂DDO-2117 的生理作用,是一种新的工具分子(图13)[64]。2019 年,有专利报道了靶向WDR5 的PROTAC分子,这项工作主要基于安大略省肿瘤研究所发现的一系列小分子。通过将分子(OICR-9429)与不同的泛素E3 连接酶底物通过各种类型的连接链连接而获得了大量PROTAC 分子。PROTAC显著地诱导了WDR5蛋白的降解,并表现出比对应小分子更好的体外抗增殖活性[65]。
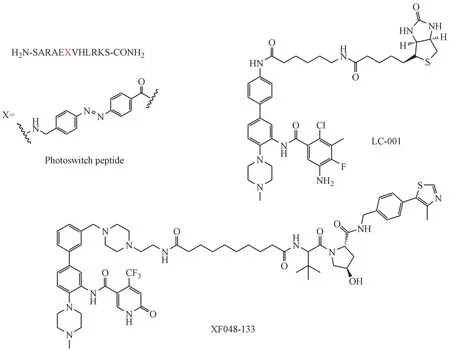
图13 基于MLL1-WDR5 PPI抑制剂的工具分子
3 总结与展望
本文对靶向MLL1-WDR5 蛋白-蛋白相互作用的小分子抑制剂的药理学机制以及WDR5 蛋白Win-site 结合位点结构信息进行了总结,介绍了已公开的各个结构类型的MLL1-WDR5 蛋白-蛋白相互作用小分子抑制剂。经过十几年的发展,人们对MLL1 的生理功能日趋了解,靶向MLL1 的新途径也不断被发现。MLL1-WDR5 蛋白-蛋白相互作用作为肿瘤治疗的新策略越来越受到研究者的关注,目前针对MLL1-WDR5 PPI 抑制剂的开发依然处于前期研究阶段,尚未有小分子抑制剂进入临床研究,但已有多个候选化合物涌现。
MLL1-WDR5 PPI抑制剂的研究虽已取得较好的进展,亦具有较大的开发潜力,但依然存在一些不足。首先,目前报道的分子虽然数量较多,但结构类型较为单一,并且无论是多肽类抑制剂还是非肽类的小分子抑制剂都含有较强的碱性基团(哌嗪、胍基和含氮芳香五元杂环等),这些结构特征可能导致分子成药性不佳(如多肽类分子体内代谢稳定性和透膜性较差、部分多取代苯基哌嗪类小分子的极性较强)。其次,部分MLL1-WDR5 PPI 抑制剂虽然具有较好的体外活性,但目前体内研究的数据较少,现有抑制剂的有效性还需要更多的实验支撑。因此,针对MLL1-WDR5 PPI 抑制剂的研究,不仅要在原有分子的基础上继续优化其成药性,还需寻找更多结构新颖的小分子抑制剂,同时抑制剂的体内有效性也有待进一步阐明。此外,利用新策略研发出更多的工具分子,可以为进一步探究MLL1 相关的生物学机制提供分子基础。
除了靶向MLL1-WDR5 蛋白-蛋白相互作用,在WDR5 蛋白上发现了另外一个蛋白结合空腔[44,66],该空腔可以介导WDR5 和RbBP5 的相互作用。研究发现,抑制该相互作用也可以抑制MLL1 甲基转移酶的活性。但该抑制剂是否确切有效暂时还未见报道,但此项研究为后续两者协同用药提供可能,为靶向MLL1 提供了更多的途径和策略。
猜你喜欢
今日农业(2022年4期)2022-11-16
分子催化(2022年1期)2022-11-02
中国农业科学(2022年14期)2022-07-26
保健与生活(2022年11期)2022-06-09
火力与指挥控制(2022年3期)2022-04-27
中草药(2022年1期)2022-01-13
保健与生活(2021年5期)2021-04-12
军民两用技术与产品(2021年10期)2021-03-16
师道·教研(2019年7期)2019-08-13
佛山陶瓷(2018年6期)2018-09-14
