
Ⅰ型神经纤维瘤病相关眼眶-眶周型丛状神经纤维瘤的临床治疗进展
2022-05-07 13:07钟民衎崔锡炜顾斌李青峰王智超
组织工程与重建外科杂志 2022年2期
钟民衎 崔锡炜 顾斌 李青峰 王智超
【提要】 Ⅰ型神经纤维瘤病是一种由nf1 基因突变导致RAS 通路调控异常的常染色体显性遗传病,其代表性特征神经纤维瘤可表现为浸润性生长的丛状神经纤维瘤。当瘤体累及眼眶及眶周组织时,可统一称为眼眶-眶周型丛状神经纤维瘤(OPPN)。OPPN 的病情复杂性为临床治疗带来了巨大挑战,现有治疗方案包括外科治疗和靶向药物治疗。外科干预手术可一定程度缓解患者情况,但整体根治效果欠佳;靶向药物治疗近年来发展迅速,部分药物临床试验也显示出不俗的研究成果。本文主要对OPPN 的机制、临床特征诊断及治疗方案进展进行综述。
Ⅰ型神经纤维瘤病(NF1)是一种较常见的常染色体显性遗传病,系抑癌基因NF1 突变导致其编码的神经纤维瘤蛋白发生功能失活而导致的多系统性病变。NF1 典型的临床症状包括咖啡牛奶斑、多发性神经纤维瘤、Lisch 结节、腋窝或腹股沟雀斑等,其中以神经纤维瘤最具代表性。约有30%~56%丛状神经纤维瘤累及眼部、眼睑、眼眶和眼眶周围等组织[1]。这些肿瘤可被称为眶颞部神经纤维瘤[2]、眶睑神经纤维瘤[3]或眼眶丛状神经纤维瘤[4]。Avery 等[5]建议将好发于这些部位的丛状神经纤维瘤统一学名为眼眶-眶周型丛状神经纤维瘤(Orbital-periorbital plexiform neurofibroma,OPPN)。OPPN 可致严重的面容畸形和功能障碍,甚至危及生命。本文主要对OPPN 的机制、临床特征诊断及治疗方案进展进行综述。
1 丛状神经纤维瘤(Plexiform neurofibromas,pNF)的发病机制
NF1 的全球发病率约为1/3 000[6],个体表达具高度可变性,约一半患者为家族性遗传突变,其余为散发型突变。NF1源于染色体17q11.2 上的NF1 基因突变,导致其编码蛋白神经纤维瘤蛋白发生功能异常[6],引起RAS 通路的异常调节及肿瘤细胞生长。pNF 被认为源于双等位基因失活NF1 的Schwann 祖细胞,主要由肿瘤Schwann 细胞、肥大细胞、神经细胞、成纤维细胞和巨噬细胞组成,而这些细胞以一种旁分泌回路方式构成一个独特的肿瘤微环境[7]。NF1-/-Schwann 细胞依靠NF1+/-神经细胞增生,透过分泌KIT 配体促进NF1+/-肥大细胞的迁移和增殖[7]。NF1+/-肥大细胞分泌TGF-beta 激活NF1+/-成纤维细胞,以致胶原合成增加,促进细胞外基质形成及NF1-/-Schwann 细胞生长[7]。这些肿瘤Schwann 细胞缺乏NF1 表达,神经纤维瘤蛋白功能失活令活性RAS 增加,激活不同的RAS 下游通路如RAF/MEK/ERK 通路和PI3K/AKT/mTOR 通路等[8],导致细胞增殖和分化失控,刺激肿瘤细胞生长,从而形成丛状神经纤维瘤。本文探讨的OPPN 正是以这种发病机制在眼眶及眶周部位呈浸润性生长的pNF。
2 OPPN 的临床特征
Ⅰ型神经纤维瘤病相关OPPN 的发病率少于10%[5]。大部分患者多于5 岁前确诊[9]。瘤体多沿三叉神经主干及其分支分布,以第一及第二分支为主[10],表现为边界不清的肿块,可触及结节,呈浸润性生长,侵袭周围组织如眼外肌、视神经鞘、眼眶深部、颞部或面颊部等。上睑下垂为最常见的临床表现,几乎发生在所有的OPPN 患者中[1,11]。OPPN 常侵犯眼睑全层组织,导致组织的局部肥大与弹性蛋白破坏,眼睑和眦肌腱松弛,加上瘤体对眼睑的机械性压迫导致上睑下垂。其他常见眼部临床表现包括眼球突出、斜视、复视、视力减弱、青光眼、角膜溃疡及睑球黏连等[11],也可同时存在牛奶咖啡斑或眼眶外神经纤维瘤等系统性临床表现。Chai 等[1]比较患者的临床表现和家族病史数据,结果显示大多数患者均表现出与患病父母或祖父母同一患侧的OPPN。有连续几代患病家族史的OPPN 患者会有更早发及更严重的临床表现。相对于长辈,他们更常伴有弱视、眼球运动障碍及眼眶骨侵袭。此外,影像学检查可发现OPPN 患者常伴有眶骨破坏、面骨及颅内侵袭等[1]。患者如伴有同侧蝶骨发育不良,影像学上可表现为蝶骨缺失或显著减小。蝶骨发育不良可致前颞叶突出到眼眶内,压迫眼外肌和视神经,造成眼球突出和斜视等临床症状。
3 OPPN 的诊断与评估
OPPN 患者在出生初期可无明显症状,幼童如伴眶周不对称或单边眼球突出,应及早进行OPPN 相关评估。临床诊疗中NF1 的诊断主要借鉴美国国立卫生研究院(NIH)于1987 年共识会议上制定的临床标准[12](表1)。2021 年,国际神经纤维瘤病诊断标准共识组(I-NF-DC)对现有NF1 诊断标准提出了修正,主要修正包括加入了基因学的诊断[13](表2)。除了NF1 临床诊断和基因检测,所有OPPN 患者应接受脑部和眼眶MRI 影像检查,利用高分辨率的MRI 平扫和增强序列来评估眼眶、面部、海绵窦等周围组织来确定诊断及评估其侵袭程度[5]。成年患者也建议进行F-FDG PET/CT 扫描来检测肿瘤恶变[14];幼年患者则应避免CT 类扫描。

表1 NF1 临床标准(美国NIH 共识会议,1987 年)

表2 修订后的NF1 临床标准(国际神经纤维瘤病诊断标准共识组,2021 年)
由于上睑下垂为OPPN 最常见的临床表现,评估上睑下垂也是OPPN 治疗方案的关键环节。多方面的术前评估有助于进一步了解患者的相关功能情况。首先,根据上睑缘遮盖角膜程度可将上睑下垂分为三级[15]:遮盖1~2 mm 为轻度,3~4 mm 为中度,大于4 mm 为重度。其次测定上睑提肌功能,按压患者眉弓处并同时观察上睑缘活动幅度,以此评估上睑提肌肌力[15]:幅度大于8 mm 为良好,4~7 mm 为中等,小于3 mm为差。除上述两项,额肌力量测定、Bell 现象及Horner 综合征检查都有助于评估患者上睑情况。
4 OPPN 的临床分型
临床上可通过各种分型来评估OPPN 的侵袭程度。早期的Jackson 分型根据眼眶受侵袭程度及视力功能受影响程度将OPPN 分为三类[16]:①眼眶软组织受累,有视力;②明显累及眼眶软组织及骨组织,有视力;③明显累及眼眶软组织及骨组织,失去视力或眼球。Lee 等[17]提出的分型则细化眶周组织受累部位,包括眉下垂型、上睑浸润伴下垂型、下睑浸润型、外眦腱断裂型、结膜及泪腺浸润型。Avery 等[5]根据OPPN的解剖位置进行了简化分类:孤立性上睑;眼睑和眶周部位;眼眶包括或不包括眼睑受累。此外,Ehara 等[18]对Ⅰ型神经纤维瘤病提出“DNB”的临床分期标准来反映严重程度,Dermatological(皮肤)、Neurological(神经)及Bone(骨骼)3 个不同系统,分别反映皮肤、神经及骨骼受累情况,并根据每个系统各自的分级(D1-D4、N0-N2 及B0-B2),总结出患病严重程度的分期,由stage1 到stage5,共5 期。根据不同分类及分期,既方便术前评估患者状况,引导后续治疗的选择,也有利于患者术后的随访观察。
5 OPPN 的治疗方案
OPPN 在颅面部呈弥漫浸润性生长,严重累及所有眶周眶内组织,且随着患者年龄增长,瘤体对组织的侵犯将进一步加重。因此,制定治疗方案前必须考虑多个因素,包括患者的年龄、视觉的发育程度、瘤体的生长速度、有否并发视神经胶质瘤、有否临床症状或功能性缺陷,以及瘤体对周边组织的侵犯程度等。如瘤体增长比同龄患者更快,应考虑OPPN恶变并安排肿瘤科会诊。整体而言,所有OPPN 患者的治疗管理决策应包含多个学科团队的意见,如整形外科、神经外科、眼科、颅面外科等[5]。
5.1 外科手术治疗
丛状神经纤维瘤一般在幼年期和青春期快速生长,此后逐渐趋于缓和[19]。这导致儿童患者与成人患者的OPPN 外科治疗方向存在一定的差异。对于成年患者,病情进展已趋缓和,传统主张进行更进取的手术以达根治性效果[17];对于儿童患者,如症状轻微,建议长期定期随访和观察;如有肿瘤所致弱视或瘤体严重侵犯眼眶及眶周软组织导致恶化的功能性缺陷和外形损坏时,应尽早行手术干预[5],才能有效避免更进一步的身体损害。Lee 等[20]报告的眼眶丛状神经纤维瘤病例中,进行最多的手术依次为肿瘤减积术、上睑下垂矫正术(前提肌切除或额肌腱膜瓣悬吊术)和外眦重建术。崔莹等[21]的回顾性系列病例研究中则主要采用三种组合术式:①肿瘤切除眼睑重建,用于上睑及外眦受累患者;②肿瘤切除联合眉下垂矫正,用于眉弓受累伴眉下垂患者;③肿瘤切除联合颅骨及眼眶重建,用于颅内组织受累或颅骨缺失致脑疝入眶内患者。对于OPPN 相关搏动性眼球突出症患者,可考虑行蝶骨缺损修补术,颅骨植骨配合钛网可用于修补蝶骨缺损及减少骨吸收[5]。
对于只有上睑下垂的患者,手术干预时机应根据下垂严重程度进行选择。视力发展受威胁的重度患者应尽早接受手术;中度患者可在合适时机进行矫正治疗;轻度患者则可先观察。治疗上睑下垂的主要手术方式包括上睑提肌缩短术、Müller 肌切除术、联合筋膜鞘悬吊术及额肌悬吊术[15]。上睑提肌缩短术及Müller 肌切除术适用于轻中度上睑下垂及提上睑肌功能良好患者[15]。提肌缩短术的优点是术后并发症较少,且符合上睑生理结构,术后外观相对其他术式更加自然。Müller 肌切除术相对简单有效,但较难矫正眼睑高度。对于提肌功能较差的重度患者,需联合上述两项术式,必要时联合筋膜鞘悬吊术,后者相对额肌悬吊术造成的并发症少但技术要求较高[15]。
然而,OPPN 的外科治疗方案面临不少挑战。首先,OPPN富含血管,在根治性手术中往往遇到术中大量出血的情况,极大增加了手术难度。其次,OPPN 无明确边界和包膜,故无法彻底切除,而残余的OPPN 瘤体也有机会继续生长并再次复发,这表示OPPN 患者就算给予外科干预手术也很难得到长远效益。基于以上原因,发展有效的药物治疗成为另一个积极探索的治疗方向。
5.2 药物治疗及临床药物试验
NF1 相关丛状神经纤维瘤的药物治疗一直鲜有突破。传统化疗不但没有明显治疗效益,还存在诱导继发恶性肿瘤的风险,例如烷基化试剂和拓扑异构酶抑制剂等[5]。近年来,随着基因组学和分子生物学技术的发展,以及对NF1 的认识逐渐加深,大量开展了针对pNF 的生物靶点研究及针对性的药物Ⅰ期、Ⅱ期临床试验,为pNF 的临床治疗带来了新的转机(表3)。稍有成效的NF1 相关pNF 临床试验包括甲磺酸伊马替尼(Imatinib Mesylate)和聚乙二醇干扰素alpha-2b(PEGIFN-a-2b)。在Robertson 等[22]开展的甲磺酸伊马替尼Ⅱ期临床试验中,发现6 例(26%)每日接受口服甲磺酸伊马替尼治疗的pNF 患者瘤体缩小达20%或以上。在Jakacki 等[23]开展的PEG-IFN-a-2b Ⅱ期临床试验中,结果显示注射PEGIFN-a-2b 可延长pNF 的肿瘤进展时间(TTP)。然而两个试验各有所限,前者样本量小(36 例)且受试者基础条件差异性较大;后者达到满意疗效的患者数量不足。
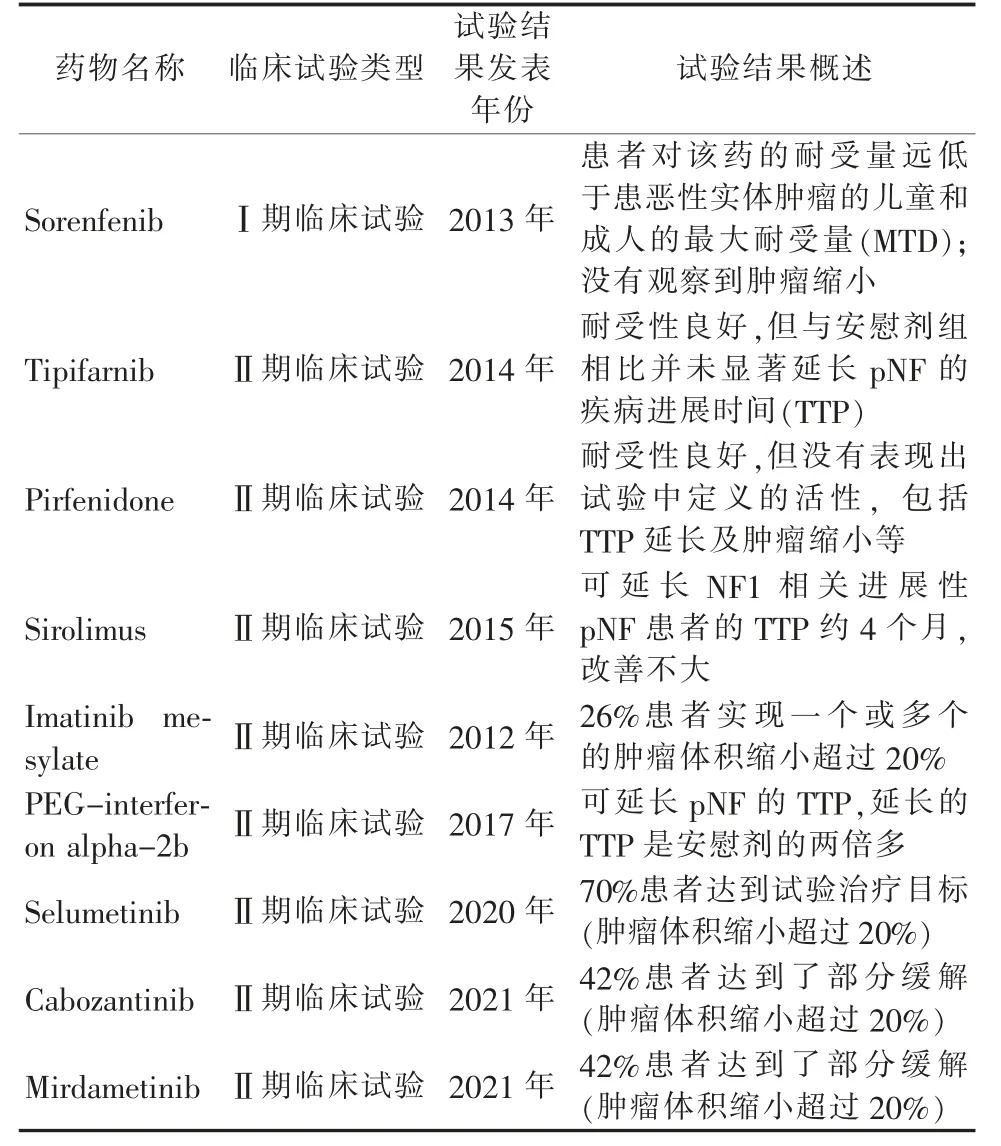
表3 近十年来主要的NF1 相关丛状神经纤维瘤临床试验列表
不少针对肿瘤细胞分子调控机制的药物试验相继出现,但都未能获得满意的治疗成果,如Kim 等[24]的Sorenfenib Ⅰ期临床试验,Widemann 等[25-26]的Tipifarnib Ⅱ期临床试验和Pirfenidone Ⅱ期临床试验,以及Weiss 等[27]的Sirolimus Ⅱ期临床试验。然而,MEK1/2 抑制剂司美替尼(Selumetinib)的成功为pNF 药物治疗带来重要突破。在Dombi 等[28]开展的Ⅰ期临床试验中,24 例pNF 幼年患者每日两次口服司美替尼,结果显示有17 例患者(71%)达到试验治疗要求(肿瘤体积缩小20%或以上)。其后,Gross 等[29]开展司美替尼的Ⅱ期临床试验,50 例幼年患者每日接受两次口服司美替尼,结果显示35例患者(70%)达到试验治疗目标(肿瘤体积缩小20%或以上),并有28 例患者表现出持续性治疗效果(持续一年或以上)。各种疼痛指标也得到显著改善。试验过程中的主要副作用包括胃肠道反应及无症状肌酸激酶升高等,但基本症状轻微。司美替尼已于2020 年4 月被美国FDA 批准用于治疗2岁及以上有症状及无法手术干预的丛状神经纤维瘤患者,这也是首个获批治疗NF1 的靶向药物。
2021 年以后,以青少年及成人患者为对象的两个NF1相关pNF 的临床试验报告相继面世,分别是卡博替尼(Cabozantinib)和Mirdametinib。卡博替尼是一种多靶点酪氨酸激酶抑制剂。在Fisher 等[30]开展的Ⅱ期临床试验中,8 例患者(42%)达到了部分缓解(MRI 评估肿瘤体积缩小20%或以上),瘤体体积变化的中位数达15.2%,无一患者在疗程期间出现病情加重,且疼痛指数也有所改善。Mirdametinib 为另一MEK 抑制剂。在Weiss 等[31]开展的Ⅱ期临床试验中,有8 例患者(42%)在12 个疗程后达到了部分缓解,仅1 名在治疗期间出现病情加重,疼痛指数方面也有显著改善。两个临床试验结果都对pNF 成年患者的治疗起到了积极作用。
6 总结
NF1 相关丛状神经纤维瘤的治疗方案长久以来都是临床上的一大难题,虽然目前手术切除是主要治疗方法,但因其病情复杂,无论是外科手术干预或传统化疗药物治疗,都未能显著解决患者的根本问题。近年来,随着分子生物学技术及靶向药物治疗的发展,丛状神经纤维瘤的治疗渐露曙光。司美替尼的成功固然为pNF 患者带来了新的希望,但其研究结果仍局限于幼年患者;新进靶向药物卡博替尼及Mir⁃dametinib 对成年患者的积极疗效或许能为成年患者带来新希望。因此,靶向治疗以及基因治疗等新的治疗手段的开发是NF1 相关丛状神经纤维瘤最值得期待的研究方向。
猜你喜欢
中华眼视光学与视觉科学杂志(2022年1期)2022-03-22
临床军医杂志(2022年2期)2022-02-25
海外星云(2021年9期)2021-10-14
现代仪器与医疗(2021年1期)2021-06-09
大众健康(2020年7期)2020-08-25
中国美容医学(2020年4期)2020-05-13
爱你(2019年13期)2019-11-14
家庭科学·新健康(2017年7期)2017-07-14
养生大世界(2016年6期)2016-06-14
