
一种TBP热解灰Pu、U分析方法
2022-05-06 07:39:12仇宏宇
中国新技术新产品 2022年3期
仇宏宇
(中核四川环保工程有限责任公司,四川 广元 610006)
0 引言
世界各国采取了很多减容效果显著的放射性废物处理工艺,焚烧法因具有减容效果显著、转型后废物的稳定性高等优点而受到各国的重视。对来自核电站、核废物处理厂的可燃废物来说,焚烧处理被证明是具有最高减容效果的技术手段。
低放有机废液(TBP/OK)经焚烧处理后转变为TBP热解灰,后期的处理处置方案与TBP热解灰的放射性水平及所含核素种类有关。其中,α核素为重点关注对象,特别是Pu、U这2种放射性核素。该文主要研究TBP热解灰中Pu、U核素的分析方法,通过调研发现,目前国内没有适用的国家标准和行业标准,大部分该行业的单位也没有建立相关的企业分析标准,相关文献资料主要集中在高放废液、后处理铀产品等方面,并没有针对TBP热解灰的分析方法。要建立一种TBP热解灰分析方法就需要解决很多难题,最大的制约因素就是浸取时焚烧灰没有完全溶解,不溶物中包括部分Pu、U核素,导致目标核素回收率不稳定;样品前处理溶液中含有大量的Ca、PO离子,会对Pu、U测量造成较大的干扰。因此,为了解决上述问题,采用微波消解、不同浓度强酸消解等方法将热解灰样品全部溶解成盐溶液,选择合适的分离提纯方法,最终形成一套合理、可行的分析TBP热解灰中Pu、U的方法。
1 分析过程
1.1 前处理
对非放射性热解灰的成分进行分析可知,其主要成分为磷酸钙、焦磷酸钙、氢氧化钙以及单质碳,并且通过成分分析发现热解灰中没有无机物。因为磷酸钙、焦磷酸钙以及氢氧化钙等均可溶解于酸,但含有约10%的碳单质,所以前期使用硝酸、盐酸浸取时无法溶解碳单质,不溶物应为碳单质。碳单质是热解时因氧气不足导致TBP氧化不充分而产生的物质,碳颗粒内可能包裹放射性核素,碳单质无法溶解就可能出现热解灰分析结果不准确的问题。由于单质碳不溶于酸,因此须采用强氧化剂对其进行氧化处理,常用的强氧化剂有浓硫酸、高氯酸等,但热解灰中钙含量较高。使用硫酸处理热解灰时,硫酸盐沉淀会对Pu、U元素进行载带共沉淀,降低Pu、U的回收率,因此选取高氯酸、浓硝酸作为氧化剂除碳,或者通过马弗炉高温加热除碳。称取TBP热解灰,在烧杯中分别加入单一浓酸和混合浓酸,从而改变混合酸配比并配合微波消解进行试验。
称取约0.2 g未被放射性污染的热解灰样品放入聚四氟乙烯烧杯中,烧杯中分别加入20 mL浓盐酸、20 mL浓硝酸以及20 mL氢氟酸,盖上烧杯盖并将烧杯置于EH20A plus型电热板上以210 ℃加热1 h后,打开烧杯盖再加热至烧杯中的液体蒸干,观察烧杯内固体残渣剩余量。
浓盐酸、浓硝酸以及氢氟酸浸煮的样品在加热过程中溶液一直呈黑色,其中浓盐酸、浓硝酸在打开烧杯盖均在30 min内蒸干,氢氟酸在加热约2 h后蒸干,蒸干后均存在较多肉眼可见的黑色残渣,溶液蒸干后在烧杯中加入5 mL硝酸,加热后黑色残渣不能溶解,溶解情况如图1所示。

图1 单一酸溶解试验情况
将盐酸、硝酸以及高氯酸相互混合,再用混合物对热解灰进行消解试验,根据不溶物的情况判断消解效果,最终选择一组合适的混合酸消解方法。
首先,选择王水(3HCL:1HNO)进行酸加入量的消解试验,判断是否因酸量不足而导致热解灰无法溶解,分别称取0.2 g未被放射性污染的热解灰样品放入烧杯中,在烧杯中分别加入15 mL盐酸、5 mL硝酸、30 mL盐酸、10 mL硝酸 、60 mL盐酸以及20 mL硝酸,盖上烧杯盖,将烧杯放在电热板上以210 ℃加热2 h后打开烧杯盖,此时溶液均呈黑色,热解灰未能完全消解。此时,在烧杯中加入5 mL高氯酸强化消解效果(继续加热),黑色逐渐褪去,溶液逐渐澄清,最终变为透明,烧杯底部均有少量白色不溶物,如图2所示。

图2 加入高氯酸前后对比
由上述试验可知,混合酸可以溶解大部分热解灰,但在加入高氯酸前,消化反应速度很缓慢,可缩短加热时间,发挥氧化作用的物质主要是高氯酸,盐酸及硝酸仅发挥辅助作用,与盐酸、硝酸的加入量的关系较小,下一步继续调整各个酸的比例并进行试验。
根据酸加入量试验结论可知,为保证试验效果,需要加入不同比例的混合酸对热解灰消解情况进行验证。
分别称取0.2 g未被放射性污染的热解灰样品放入烧杯中, 编号为1#、2#、3#、4#和5#,根据表1分别在烧杯中加入混合酸, 在电热板上以210 ℃进行加热至溶液蒸干,待溶液冷却后加入5 mL硝酸,将残渣溶解后,观察溶液内不溶物的情况。

表1 混合酸加入情况
完成反应后,各组均有白色不溶物。其中,2#、3#以及4#不溶物少于1#、5#,试验说明盐酸、硝酸以及高氯酸组合消解效果比高氯酸与盐酸、硝酸单独组合的消解效果好。
分别称取0.2 g未被放射性污染的热解灰样品放入烧杯中, 编号为1#、2#和3#,根据表2分别在烧杯中加入混合酸, 在电热板上以210 ℃加热至溶液近干,待溶液冷却后加入5 mL硝酸,将残渣溶解后观察溶液内不溶物的情况。

表2 混合酸加入情况
试验各组热解灰均全部溶解。其中, 2#、3#、7#以及8#试验的不溶物较少。混酸可以将热解灰基本溶解,仅剩余极少的白色细小不溶物,如图3所示。

图3 溶解效果对比
继续改变混合酸的比例并进行试验,混合酸比例见表3。

表3 混合酸加入情况
其中,6#样品剩余的不溶物较少,重复溶解1次后就可以全部溶解。
使用消解常用的硝酸、氢氟酸以及盐酸进行消解试验。
称取约0.2 g未被放射性污染的热解灰样品放入消解罐中,在消解罐中分别加入15 mL浓盐酸、15 mL浓硝酸以及15 mL氢氟酸, 使用WX-6000微波消解仪对样品进行消解,消解条件为在160 ℃的环境下消解 2 h。消解结束后溶液呈淡黄色,底部均有少量不溶物。
该试验使用WX-6000微波消解仪对样品进行消解,高氯酸在微波消解过程中存在危险,该设备使用说明中严禁加入高氯酸,并且消解所加酸不超过15 mL,每次消解样品量不超过0.2 g,因此按不同比例混合硝酸、盐酸并进行微波消解试验。
本次报告显示,手术切除和注射碘酊治疗口腔粘液腺囊肿治愈率均达到95%以上,临床疗效确切,两种治疗方法没有显著性差异。10例复发病例(手术切除治疗4例,注射碘酊治疗6例),其原因可能是囊壁处理不彻底,部分囊壁残留导致囊肿复发。
在设备推荐条件下,使用不同的混合酸比例对试验条件进行初选。称取约0.2 g未被放射性污染的热解灰样品放入消解罐中,编号1~编号6所加混合酸比例见表4,消解条件为在160 ℃的环境中消解2 h。消解结束后溶液呈淡黄色,溶液略浑浊,加热蒸干后,溶液变为透明,有少量白色不溶物。其中,1#、4#以及6#热解灰中不溶物较少。其中,2#、4#以及6#的不溶物含量明显小于1#、3#以及5#,因此选择比例为 1∶1、2∶1以及1∶2的硝酸、盐酸进行下一步试验。
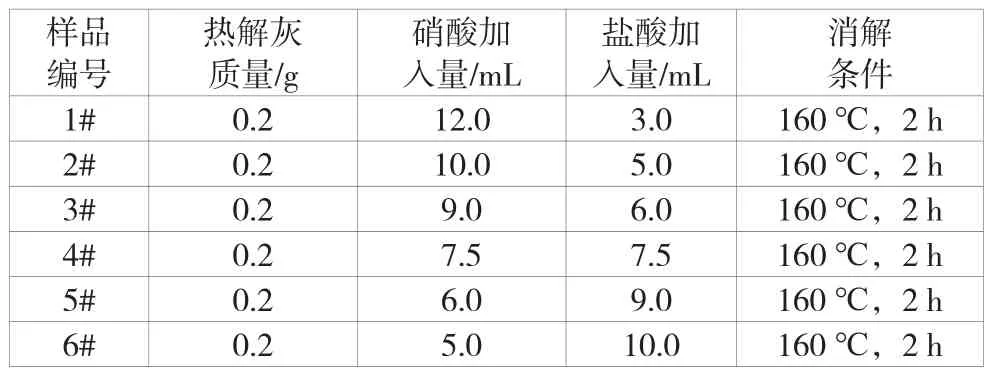
表4 微波消解情况
称取约0.2 g未被放射性污染的热解灰样品放入消解罐中,编号1~3所加混合酸比例见表5,消解条件为在190 ℃的环境中消解2 h。消解结束后溶液呈淡黄色,溶液略浑浊,加热蒸干后,溶液变澄清透明,3组试验最终溶液仍含有少量白色不溶物 ,不溶物含量相近,但不溶物均少于试验一中不溶物的量。

表5 微波消解情况
分别称取0.1 g未被放射性污染的热解灰样品放入消解罐中,编号1~3所加混合酸比例见表6,消解条件为在190 ℃的环境中消解2 h。消解结束后溶液呈淡黄色,溶液略浑浊,加热蒸干后,溶液变为透明,该试验的最终溶液中仍含有少量白色不溶物 ,但不溶物均少于试验一中不溶物的量。

表6 微波消解情况
该试验1#、4#样品中无可见不溶物,其他样品中含有少量不溶物,4#样品消解情况如图4所示。

图4 微波消解情况试验
1.2 分离提纯
根据选定的微波消解前处理方案,制备6组前处理溶液,编号为A-1、A-2、A-3、B-1、B-2和B-3, 在前处理液中加入1 mL的10 ng/mL铀标准溶液。
充分搅拌溶液,使前处理液与加入的铀标准溶液混合均匀,在制备的前处理溶液中加入4 mol/L的氢氧化钠溶液,将溶液的pH值分别调为10、11和12,溶液充分沉淀后将其离心分离,并收集清液。使用3 mol/L的硝酸将清液的pH值调为3,使用pH值为3的硝酸将清液转移至100 mL的容量瓶中,并使用分光光度法、激光荧光法对其进行测量。
取5个100 mL的容量瓶,分别加入1.0 mL、2.5 mL、5.0 mL、10.0 mL以及25.0mL的100 ng/mL铀标准溶液,在容量瓶中加入2滴10%乙二胺四乙酸溶液、2滴2 mol/L的酒石酸溶液以及1滴3 mol/L的硝酸并摇匀,再加入2 mL的0.02%偶氮胂III,使用pH值为3的醋酸氯缓冲溶液定容至刻度线,充分摇匀后静置5 min;将标准溶液转移至10 mm的比色皿中,将分光光度计的波长调为650 mm,以空白试剂为参比溶液,依次测量各标准溶液的吸光度值,并绘制校准曲线,曲线相关系数大于或等于0.999。
在A-1、A-2以及A-3容量瓶中加入2滴10%的EDTA溶液、2滴2 mol/L的酒石酸溶液以及1滴3 mol/L的硝酸并摇匀,再加入2 mL的0.02%偶氮胂III,使用pH值为3的醋酸氯缓冲溶液定容至刻度线,充分摇匀后静置5 min;将标液转移至10 mm的比色皿中,将分光光度计的波长调为650 mm并进行测量。
取定容好的A-1、A-2和A-3样品,使用微量测铀仪进行激光荧光法测量,样品加标回收率同样不满足分析需求。
在B-1、B-2以及B-3前处理液中分别加入1 mL的100 ng/mL铀标准溶液,使用2 mol/L的硝酸定容,并开展TBP萃取试验:1) 准确量取4.0 mL试样放入15 mL的萃取管中,加入1~2滴氨基磺酸亚铁溶液,摇匀后反应5 min,再加入1.0 mL盐析剂摇匀。2) 加入5.0 mL萃取剂萃取3 min,离心1 min后分相,弃去水相。3) 在有机相中加入1.0 mL水,反萃5 min,离心1 min分相,收集水样待测。4) 继续在有机相中加入1.0 mL的5%NaCO溶液,反萃3 min,离心1 min后分相,收集并合并水相。使用分光光度法、激光荧光法对提纯效果进行验证。
在前处理液中加入1mL的2.42 Bq/mL钚标准溶液,使用2 mol/L的硝酸进行定容,在不同浓度的硝酸介质中进行TIOA萃取试验:1) 取10 mL样品,加热至近干,使用pH值为2的硝酸溶解沉淀,使用约5 mL不同酸度的硝酸将溶液转移至梨形分液漏斗中,加入6滴氨基磺酸亚铁溶液直到不产生大量气泡,摇匀放置10 min。2) 加入6滴0.2 mol/L亚硝酸钠溶液至大量气泡消失,放置10 min。3) 加入4.0 mL TiOA溶液,在振荡器中震荡3 min,去除水相,取有机相至离心管,离心分相3 min,取有机相3.0 mL,加10.0 mL闪烁液并摇匀,使用液闪谱仪进行测量。
2 试验结果及讨论
2.1 确定前处理的方式
由于浸取法无法得到稳定的回收率,因此采用能够将TBP热解灰样品全溶解的方法进行前处理,土壤沉积质和植物等固体环境样品中钚的定量分析大都包括样品消解、分离纯化和仪器测量等步骤,样品消解是其中至关重要也是耗时最长的步骤。单一酸均未能全部溶解TBP热解灰样品,由于TBP热解灰样品放射性活度较高,因此也应该考虑前处理的耗时问题,通过对比筛查(表7),最终确定前处理方法为使用硝酸、盐酸混合酸进行微波消解。

表7 几种前处理方式的特点对比
2.2 确定铀分析方法
常采用激光荧光法对样品铀进行分析,TBP热解灰样品前处理溶液中钙离子含量较高,须对铀加标样品进行干扰去除处理后再使用微量测铀仪进行分析。该试验采用沉淀提纯和萃取提纯2种干扰去除方法,当使用沉淀提纯方法时,氢氧化钙沉淀载带了样品中的全部铀元素,无法分离铀和钙元素。当使用TBP进行铀萃取时,可以达到较好的效果,具体结果见表8。

表8 不同干扰去除方法铀回收率情况
根据表8对比干扰去除方法和铀回收率情况,在同样的加标情况下,使用TBP萃取2次铀的回收率最高且相对标准偏差较低,由此确定铀的分析方法为使用TBP萃取2次后采用微量测铀仪进行分析。
2.3 确定钚分析方法
用液体阴离子交换剂TIOA分离钚有许多优点,该文主要研究TIOA从硝酸介质中萃取钚的条件,分别使用酸度为3.0 mol/L、3.5 mol/L、4.0 mol/L、4.5 mol/L以及5.0 mol/L的硝酸对加标样品的钚回收率进行测定。酸度萃取时钚回收率见表9。
根据表9对比样品酸度和钚回收率情况,在同样的加标情况下,使用TIOA萃取时,酸度为4.5 mol/L时钚回收率最高,由此确定采用4.5 mol/L硝酸介质对钚进行TIOA萃取分离。
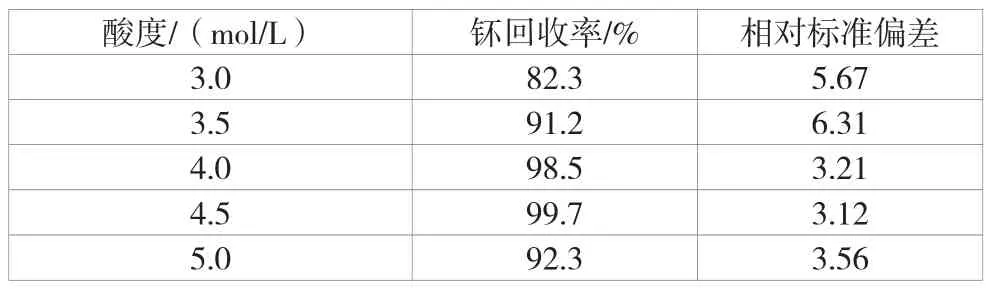
表9 酸度萃取时钚回收率情况
2.4 方法比对验证
在确定了TBP中Pu、U分析方法后,组织核工业203研究所、中核四川环保工程有限责任公司两家单位进行方法比对,以验证方法的精密度。采集满足分析要求的热解灰样品,分成2份平行试样,每份试样3 g~5 g,对给定的分析方法进行验证,每份试样进行6个平行试样测定。TBP热解灰样品Pu、U实验室间分析结果平均值间偏差均小于10%,方法精密度、加标回收率验证结果汇总表见表10。

表10 实验室间比对情况
根据表10可以得出以下结论:1) 2家实验室对TBP热解灰中Pu分析方法的精密度验证表明,实验室内相对标准偏差为3.21%~3.47%,样品加标回收率为93.76%~99.40%,说明TBP热解灰中Pu分析方法的精密度及加标回收率较好。2) 2家实验室对TBP热解灰中U分析方法的精密度验证表明,实验室内相对标准偏差为2.66%~3.12%,样品加标回收率为86.24%~99.70%,说明TBP热解灰中U分析方法的精密度及加标回收率较好。
3 结论
该文建立了一种TBP热解灰Pu、U分析方法—利用浓硝酸、浓盐酸进行微波消解的方法,将TBP热解灰样品全部溶解,结合TBP热解灰成分特点,采用TBP萃取-激光荧光法、TIOA萃取-液闪测量法对铀、钚进行分析,解决了TBP热解灰回收率低以及样品中干扰离子影响分析的问题,该方法回收率大于95%,相对标准偏差小于5%,为TBP废液焚烧项目运行及TBP热解灰后续处理提供了技术支持。
猜你喜欢
江南诗(2023年6期)2023-12-08 05:17:24
发明与创新·小学生(2022年12期)2022-12-09 22:54:54
金沙江文艺(2022年1期)2022-02-04 10:15:16
北京航空航天大学学报(2021年9期)2021-11-02 08:24:32
化学教与学(2021年12期)2021-02-18 01:16:58
中学生数理化(高中版.高考理化)(2020年1期)2020-11-24 21:02:17
重型机械(2020年2期)2020-07-24 08:16:16
中国航海(2019年2期)2019-07-24 08:26:40
红领巾·探索(2014年7期)2014-10-10 02:13:56
中学生数理化·八年级物理人教版(2014年2期)2014-04-02 09:49:04
