
肯尼迪病3例并文献复习
2022-04-28 01:29董思语史兆春金善泉秦晓旋
罕少疾病杂志 2022年5期
孙 美 董思语 刘 东 史兆春 金善泉 秦晓旋
南京医科大学第一附属医院神经内科 (江苏 南京 210029)
肯尼迪病(Kennedy's disease,KD),又称脊髓延髓肌肉萎缩症,是一种由雄激素受体(androgen receptor,AR)基因第一外显子CAG异常扩展导致的X连锁隐性遗传的神经肌肉病。KD患者多在30~40岁发病,以脊髓、延髓肌无力,雄激素不敏感等为主要表现,该病进展缓慢,确诊多年后仍可保持良好的运动功能,10年生存率可达82%[1]。KD误诊率高,易与运动神经元病等疾病相混淆。本文拟通过对我院基因确诊的3例KD患者的临床资料进行分析,结合相关文献复习,对该病的临床表现、发病机制、治疗预后等方面加以论述、讨论,旨在提高临床医师对该病的认知。
1 临床资料
1.1 病例1男性,31岁,因“双下肢乏力1年,双小腿疼痛3月余”于2020年1月13日收住我院风湿科。患者2019年1月出现双下肢乏力,跑步等高强度运动不能耐受。2019年9月出现双小腿肌肉抽痛,运动后加剧,上楼等动作费力。2019年12月查肌酸激酶:1189.9U/L,门诊拟以“肌炎”收住风湿科病房。患者育有1子,为试管婴儿,患者外公有类似症状。其余既往史、个人史及家族史无特殊。查体:神清,精神可,乳腺发育,心肺腹查体(-)。专科查体:高级智能检查正常,颅神经查体(-),双手震颤,肩胛带肌及下肢近端肌肉萎缩,四肢近端肌力4-级,远端肌力5级,四肢腱反射(+),病理征(-),深浅感觉未见异常。入院后实验室检查:肌酸激酶:1098U/L↑,风湿免疫相关抗体(抗ENA抗体、自身免疫性肌炎抗体检测、抗核抗体)、甲状腺功能、肝肾功能电解质均正常。肌电图:上下肢神经源性损害(慢性失神经损害为主,偶可见自发电位)。肌肉活检:符合神经源性病理改变。基因检测:AR基因CAG重复次数为51次(图1)。

图1 病例1患者基因检测图
1.2 病例2男性,53岁,因“四肢乏力、麻木3年,加重6月”于2020年6月13日收住于我院风湿科。患者3年开始逐渐出现双下肢乏力,进行性加重,当地医院考虑“腰椎间盘突出”,牵引等物理治疗后未见好转,并逐渐出现四肢麻木感。6个月前,患者症状明显加重,活动困难。门诊拟以“皮肌炎”收住风湿科病房。患者既往有“高血压”病史,有“阑尾炎、痔疮”手术史,有“青霉素”类药物过敏史,有长期酗酒史20年。患者育有1女,为自然生产,其他既往史、个人史及家族史无特殊。查体:神清,精神可,双侧乳房发育,心肺腹查体(-)。专科查体:高级智能检查正常,颅神经查体(-),口周及双手静止性震颤,未见明显肌肉萎缩,四肢近端肌力4级,远端肌力5-级,四肢腱反射(-),病理征(-)。深浅感觉未见异常。实验室检查:肌酸激酶:565U/L↑,甘油三脂:3.84mmol/L↑,肿瘤标志物、甲状腺功能、抗ENA抗体、抗核抗体肝肾功能电解质无异常。T细胞亚群:CD3+CD4+(辅助T细胞):48↑%,CD3+CD8+(抑制性/细胞毒T细胞):14.5%↓。CD3(总T淋巴细胞):62.3%↓,CD19(B细胞):21%↑。性激素(FSH+LH+PRL+E2+T+P) :睾酮:11.73nmol/L↓,孕酮:0.6nmol/L↓,泌乳素:106.19mIU/L↑。肌电图:广泛神经源性损害(慢性失神经损害),多处神经可见感觉纤维传导波幅下降或测不出。肌肉活检:符合神经源性病理改变。基因检测:AR基因CAG重复次数为49次(图2)。
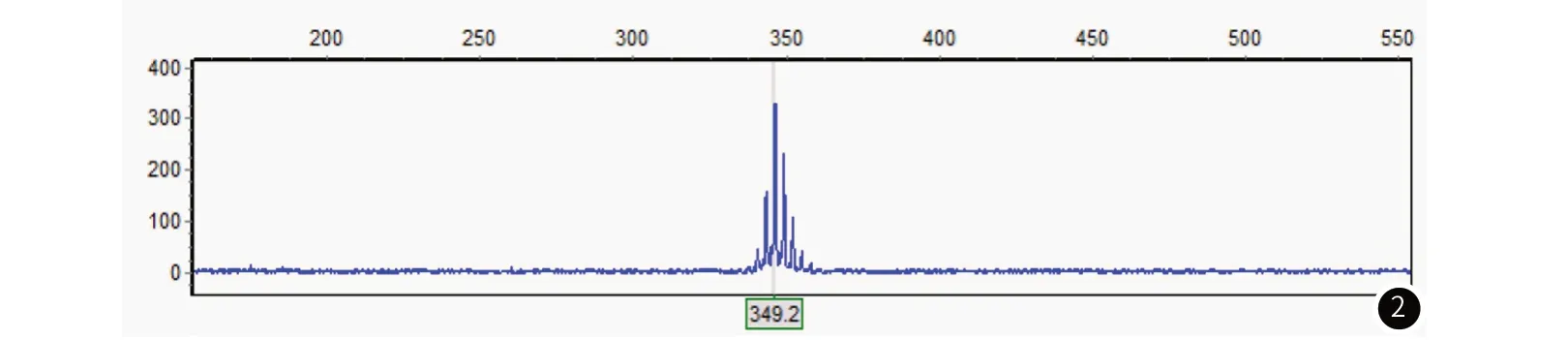
图2 病例2患者基因检测图。
1.3 病例3男性,58岁,因“双下肢无力6年,加重1年”于2020年10月13日收住于我院神经内科。患者6年前无明显诱因出现双下肢无力,蹲下起身稍费力,行走较长时间感小腿肌肉酸痛,未在意。近1年来无力加重,上坡困难,容易摔倒,并自觉双上肢精细活动差,提重物较长时间后左上肢体乏力明显,偶尔饮水呛咳、流涎。患者有“颈部脓肿切开引流术”史,育有1子1女(均为正常生产),但性功能障碍多年。其余既往史、个人史及家族史无特殊。查体:神清,精神可,心肺复查体(-)。专科查体:高级智能检查正常,颅神经查体:舌肌萎缩、纤颤。余肢体未见明显萎缩,四肢近端肌力4级,远端肌力5级,双上肢腱反射(-),双下肢腱反射(+),病理征(-)。四肢深浅感觉正常。实验室检查:生化全套:乳酸脱氢酶:272U/L↑,肌酸激酶:438U/L↑,甘油三脂:4.67mmol/L↑。性激素(FSH+LH+PRL+E2+T+P) :泌乳素:309.72mIU/L↑,雌二醇:202pmol/L↑。肝肾功能电解质、肿瘤标志物、抗ENA抗体、抗核抗体、尿轻链、甲状腺功能未见明显异常。肌电图: 广泛慢性神经源性损害,所测神经感觉纤维传导波幅下降或测不出。基因检测:AR基因CAG重复次数为45次(图3)。本研究病例的基本情况和临床表现见表1。
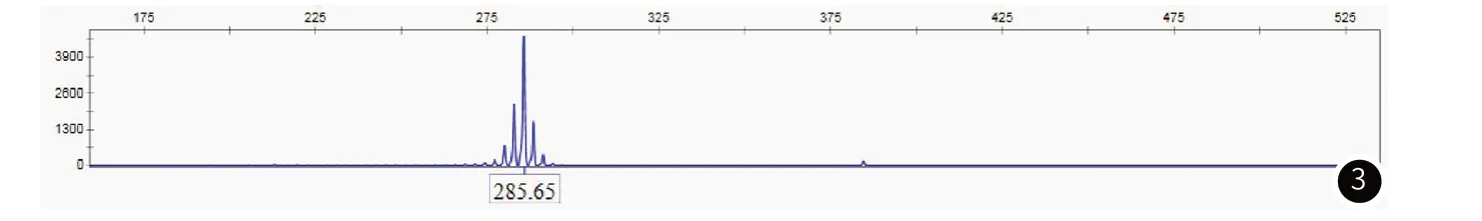
图3 病例3患者基因检测图。
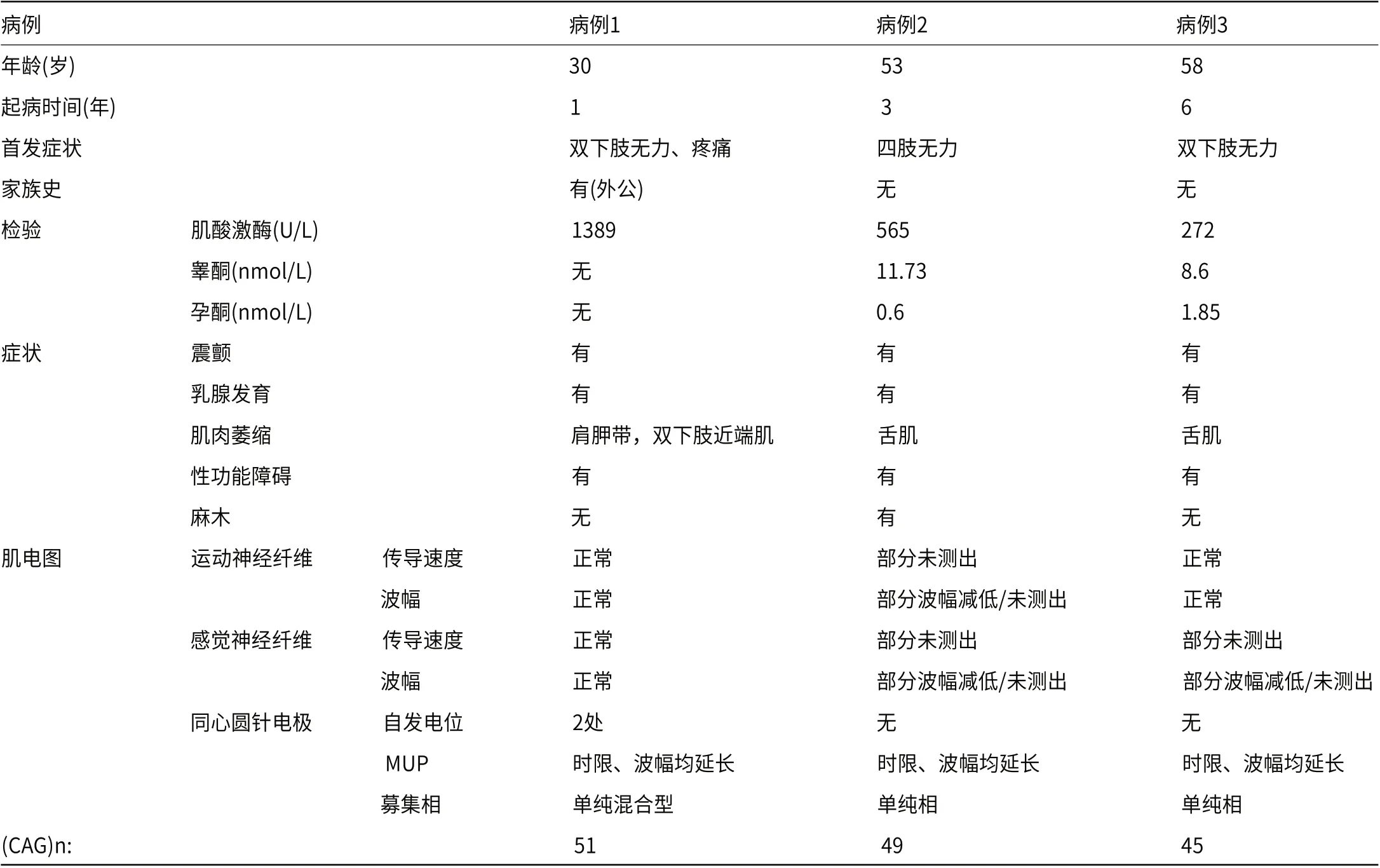
表1 3例肯尼迪患者的临床特征
2 讨 论
KD由Kennedy等[2]在1968年首先报道,该报道详细描述了11位来源于两个家系的缓慢进展的脊髓延髓肌无力患者,并提出KD很大可能为一种X连锁的隐性遗传性疾病。1991年La Spada等[3]提出KD的致病基因是位于X染色体q11-12位置上的AR基因,由CAG异常扩增导致。KD发病率低,世界范围内均有病例报道,但缺乏大型的流行病学调查,现有的小范围调研估计KD的发病率在1.58/100000~15/100000之间[4-6],但考虑到KD主要为男性患者,女性携带者甚至是女性纯合子患者可没有症状或者症状非常轻微,KD的发病率可能被严重低估。
AR归属于类固醇/甲状腺激素受体超家族,非激活状态时与热休克蛋白等结合组成复合体,一旦与睾酮或二氢睾酮结合激活,可与热休克蛋白分离并发生构象改变进入细胞核内,后经核移位、染色质结合、与转录辅助因子的相互作用调节基因表达。AR对男性性分化及青春期性发育起重要作用,在肾脏、骨骼肌、肾上腺、皮肤和中枢神经系统等非生殖器官中也均有表达,这表明AR对各级组织均有重要影响[7-8]。CAG异常扩增可导致多聚谷氨酰胺延长,AR转录活性抑制,而动物实验证实,AR异常表达对KD的发生发展至关重要[9-10],可能与AR功能丧失导致神经元变性和异常AR的神经元毒性作用相关[11]。此外,动物实验还证实降低血清睾酮可阻止KD疾病症状的出现,这也从一定程度上说明KD是一种激素依赖性疾病[12]。
KD可出现全身多系统受累的症状体征。包括神经系统症状,神经心理异常、激素水平紊乱、代谢改变、心脏、骨骼及生殖泌尿系统改变等。Atsuta等[13]的研究纳入了223名KD患者,是迄今为止纳入病人数最多的研究,该研究重点研究KD病程中的9个里程碑事件,分别是手部震颤、肌肉无力、爬楼困难(扶手上楼梯)、构音障碍、吞咽困难、使用拐杖、使用轮椅、吸入性肺炎、肺炎后死亡死亡,患者出现上述事件的中位年龄分别为33岁、44岁、49岁、50岁、54岁、59岁、61岁、65岁,且CAG重复次数与里程碑事件的发生年龄患者显著负相关,但与疾病进展速度无关。
一项针对我国东部地区的KD患者研究显示,35.5%的KD患者出现球部症状,96.1%出现肢体无力(75.5%出现下肢无力),34.8%出现男性乳房发育,13.5%出现手部震颤[14]。KD患者内分泌受累可先于神经系统症状之前出现,雌激素水平增高,睾酮水平异常(增高或降低)[15],这可能与不同患者所处年龄及疾病进展阶段不同有关。如本文结果所示,三例病患均有震颤、肢体无力、乳房发育,病例2和病例3的睾酮水平均降低,雌激素水平正常。电生理检查是KD诊断的重要环节,对多种亚临床病变区域的检测更为敏感,将周围神经以延髓段、颈段、胸短、腰骶段进行划分,则KD的神经电生理异常可表现为多个节段的慢性进行性神经源性损害,易与运动神经元病混淆,但KD也易累及感觉神经纤维,如本文所述,两位患者均出现感觉纤维传导的波幅度减低。
综上所述,KD是一种罕见而复杂的全身性疾病,以神经、内分泌、肌肉受累症状为主,详细的病史采集、体格检查、实验室检查、基因检测有助于疾病的早期诊断。KD尚缺乏有效治疗手段,目前只能根据临床症状进行相应的对症处理,本研究期待未来的多中心合作,为疾病的基因治疗、生物学标志物等方便提供更多线索。
猜你喜欢
康复(2022年25期)2022-11-18
中国现代药物应用(2022年17期)2022-10-17
中国现代医生(2022年21期)2022-08-22
现代仪器与医疗(2022年2期)2022-08-11
农民致富之友(2018年13期)2018-07-13
中国医药科学(2017年9期)2017-08-04
东方教育(2016年15期)2017-01-16
中国民族民间医药·下半月(2016年4期)2016-05-24
审计与理财(2014年8期)2014-10-16
中国民族民间医药·下半月(2014年2期)2014-09-26
