
Coronary Artery Calcification and The Roles of Related Chinese Herbal Medicine
2022-04-24 06:15TIANZhiZHANGYuanLIZhengGong
生物化学与生物物理进展 2022年4期
TIAN Zhi,ZHANG Yuan,LI Zheng-Gong
(Chongqing General Hospital,Department of Cardiology,University of Chinese Academy of Sciences Chongqing Hospital,Chongqing 400013,China)
Abstract Artery calcification(AC)is a pathological phenomenon in the process of atherosclerosis,especially the coronary artery calcification (CAC) of the arterial intima. Originally, CAC is a self-protection mechanism to reduce atherosclerotic inflammation.However, it also acts as a significant cause of atherosclerotic plaque rupture, particularly of the microcalcification in the early stage of CAC, which is the leading cause of plaque rupture. From the microcalcification in the early stage to the stable fusion in the late stage, different degrees of CAC have different effects on cardiovascular events. Chinese herbal medicine (CHM) has been used to treat atherosclerotic cardiovascular diseases for centuries.How CHM is effective in the treatment and what mechanism is involved in CAC need further investigation. Here, we review the molecular mechanisms of the development of CAC and the influence of CHM on those pathological processes.
Key words atherosclerosis,coronary artery calcification,Chinese herbal medicine,pharmacology,drug therapy
Artery calcification (AC) is a complication in the development of artery atherosclerosis (AS). Coronary artery calcification (CAC) is the calcification of the arterial intima. It is the ectopic deposition of calcium phosphate in vascular intima and is also a significant risk factor for coronary artery disease (CAD)[1-2]. In addition to traditional cardiovascular risk factors,CAC can also provide additional predictive information for major adverse cardiovascular events(MACE)[3]. CAC is one of the critical causes of coronary plaque rupture. Intervention in the pathological process of CAC can help to reduce the risk of plaque rupture and to improve the clinical prognosis of patients with CAD. Chinese herbal medicine (CHM) has a long history of treating atherosclerotic cardiovascular disease (ASCVD), with reasonable effectiveness and safety.What impact does CHM have on CAC? Is there any reference for modern intervention in CAC? Therefore, we review the pathological process of CAC and the mechanism of action of CHM on CAC.
1 The mechanism of CAC
1.1 Epidemiology
CAC is an essential symbol of possible coronary artery events and a significant risk factor for atherosclerosis complications. It is closely related to the risk of ASCVD events within ten years[4]. It is estimated that double CAC scores increase the risk of ASCVD events by 14%.
AC can occur in the arterial intima or media.CAC occurs in the arterial intima, which can cause plaque to rupture easily.Arterial media calcification is more common in peripheral arteries of extremities[5].There are racial differences in the degree of CAC,which may be the basis for differences in clinical outcomes among different ethnic groups. CAC is heavier in the Whites and is lower in African Americans[6].
1.2 The developmental process of CAC
Atherosclerosis began when lipoproteins invaded the vascular intima[7], causing local inflammation and releasing chemotactic peptides. Monocytes are attracted to the intima, enter the vascular tissue, and transform into macrophages. Macrophages engulf and break down lipoprotein cholesterol complexes. In the process of catabolism, the lipoprotein cholesterol complex is oxidized. Oxidized cholesterol has a toxic effect on macrophages, leading to apoptosis and necrosis of lipid-containing macrophages(foam cells).Pro-inflammatory factors and toxic substances released by necrotic macrophages further enhance the inflammatory response[8].
There are two different processes in bone development. One is directly transforming connective tissue into bone, which is called intramembranous bone formation. The other is indirectly replacing the originally formed cartilage matrix by bone, which is called endochondral bone formation. CAC is similar to intramembranous bone formation[9].Intramembranous bone formation begins with the differentiation of mesenchyme cells into osteoblasts,secreting the organic extracellular bone matrix.Transcription factors core-binding factor α1 and osterix (OSX) play essential roles in regulating this differentiation process[10].
Macrophages and osteoblasts release matrix vesicles (MVs), where hydroxyapatite crystals are deposited and located in the extracellular matrix[11]and involved in the mineralization of the bone matrix.By accumulating calcium and phosphate, crystals grow in MVs, deposit on collagen. Finally,hydroxyapatite crystals penetrate the bilayer of MVs.The extraosseous crystals gradually increase and become calcified particles called microcalcification[12].These tiny calcium particles will further fuse, expand, and form more extensive platelike calcium deposits called calcified nodules[13].Ultrathin section analysis of plaques found that MVs in the fibrous caps of unstable plaques were significantly more than those in stable plaques. There were more calcium deposits, which indicates the promotion of MVs in CAC[14]. During the progression of atherosclerosis, macrophages underwent apoptosis,inflammatory and cell death; MVs become the place for calcium complex crystallization; the expression of calcification inhibitors are less; and more vascular smooth muscle cells (VSMCs) differentiate into osteoblast-like cells. These four are the initiation mechanisms leading to early CAC[15].
1.3 Significance of CAC
AC process has a positive meaning. With the development of calcification, it can control the spread of inflammation and physically protect adjacent tissues from damage. In a sub-study with data from the multi-ethnic study of atherosclerosis(MESA)[16],it was found that CAC volume was positively correlated with ASCVD risk. But at the same level of CAC volume, CAC density was significantly negatively correlated with MACE risk. Other studies comparing acute and stable CAD also showed that denser calcified plaques are associated with stable CAD[17].Clinical trials of statin therapy have found that CAC in the statin group progressed faster[18]. Also, to narrow down the lipid core of unstable plaques,statins have a beneficial effect on ASCVD risk. The calcium density of plaques enhances the fibrous cap’s stability[19]and is more conducive to ameliorate the prognosis of patients. Statins can reduce the risk of cardiovascular events in CAC patients, but they do not affect people with CAC score of 0[20]. Another study[21]found that the incidence of cardiovascular events was 5.5%, 22.7%, and 37.7% in patients with only calcified plaques, patients with single noncalcified plaques and patients with both calcified and non-calcified plaques, respectively. These results indicate that calcified plaques are more stable and have fewer cardiovascular events.
From an anatomical analysis, ruptured plaques have higher circumferential stress than regular arterial segments[22]. Destroy stress usually occurs at the interface between materials with different stiffness.Since the hardness of calcified plaques is 4-5 times more than that of cellular plaques[23], the damage stress is concentrated at the interface between calcified and non-calcified areas in the plaques[24].With the progress of CAC, the number of interfaces between calcified and non-calcified areas will initially increase. After further calcifications merge, the calcified interface area will decrease. Therefore, the risk of plaque rupture initially increases with increasing calcification. However, with the decreasing of calcified interface area and damage stress of the plaques, the risk of rupture of calcified plaques decreases. Therefore, microcalcification in the early CAC stage is an essential factor leading to plaque instability[25].
1.4 Calcification stages
A study of observing microcalcifications in human coronary arteries with high-resolution microcomputed tomographe (1172, 2.1 μm resolution,SkyScan, Belgium) found that microcalcifications>5 μm can transform stable plaques into vulnerable plaques[26]. Microcalcifications <5 μm do not affect the stability of atherosclerotic plaques because the tiny voids associated with these very small particles limit the growth of microcalcifications due to their surface energy[27-28]. Previous studies have found that early spotty calcification (less than 1 000 μm) may be related to plaque rupture[29].A recent study on carotid plaques also mentioned that microcalcification is associated with plaque instability, while large calcification increases plaque stability[30].Microcalcification was defined in this study≤500 μm,and macrocalcification>500 μm. The relationship between the diameter of calcified arterial nodules and clinical events still requires further investigation.Based on the above findings, we roughly divide CAC progression into three stages: the early stage is microcalcification diameter<5 μm, microcalcification during this period does not cause plaque instability;the middle stage is calcified nodules diameter 5-500 μm, during this period, the stress between calcified nodules and non-calcified areas increases,which is the time when atherosclerotic plaques are most likely to rupture; the late stage is macrocalcification diameter >500 μm (Figure 1). Due to the mutual fusion of calcified nodules in the late stage, the contact area between calcified and noncalcified areas is reduced, the damage stress of calcified nodules is reduced, and the risk of plaque rupture is reduced. Statins can reduce the risk of plaque rupture by promoting plaque calcification[31]and reduce cardiovascular events.
In the early stage of CAC, the apoptosis of macrophages, the formation of MVs, the reduction of calcification inhibitors, and the differentiation of osteoblasts are all factors that initiate calcification.Therefore, theoretically, methods that can inhibit macrophage apoptosis, atherosclerotic inflammation,and osteoblast differentiation can inhibit CAC. When the microcalcification diameter is 5-500 µm, because the contact area between calcified area and noncalcified area increases, the damage stress in atherosclerotic plaque increases, and this period is the time when the risk of plaque rupture is the highest.As the microcalcifications further develop and fuse into larger calcified nodules, the risk of plaque rupture decreases. Thus, methods that reduce the calcified interface area by promoting calcified fusion may contribute to calcified plaque stabilization. In the late stage of CAC, stable calcified lesions will not increase the risk of plaque rupture. Previous evaluation of vulnerable plaques referred to lipid pool,necrotic core, fibrous cap thickness, and other indicators. According to the current study, the diameter of calcified nodules in plaques are also an important indicator leading to plaque vulnerability.PET in conjunction with18F-sodium fluoride (18FNaF) has been demonstrated to identify active calcification and/or microcalcifications of the CAD process[31]. Further studies may be able to estimate plaque vulnerability by scoring the plaque vulnerability markers.
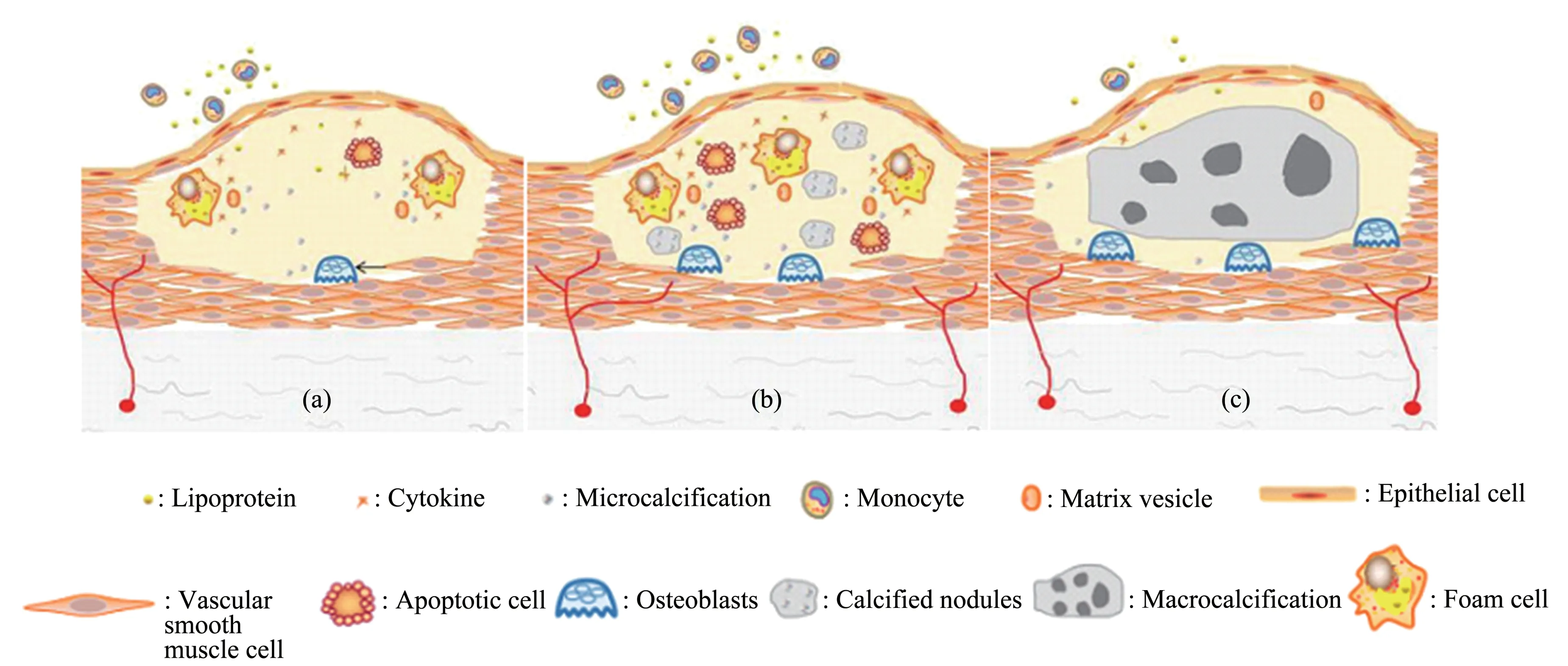
Fig.1 Calcification stages
1.5 The promoting and inhibiting factors of CAC
1.5.1 Promoting factors
After incubating calcified vascular cells with tumor necrosis factor-α (TNF-α). Calcium deposition increased in a dose-dependent manner. In human VSMCs exposed to calcification mediators, TNF-α also enhanced the expression of core-binding factor α1, osterix and alkaline phosphatase (ALP), and bone sialoprotein(BSP)[32].
Bone morphogenetic protein (BMP) is a highly effective osteoinductive factor that can induce cells derived from non-bone tissues to differentiate into osteoblasts, leading to ectopic bone formation. BMP has a tremendous ability to initiate bone formation[33].
Metabolic disorders produced by diabetes and chronic kidney disease (CKD) affect the homeostasis of calcium and phosphate in blood vessel walls.These patients have a higher rate of CAC. Fibroblast growth factor(FGF)23 is released from bone cells and regulates the elimination of phosphate[34]. However,there are still reports suggesting that the biomedical effects of FGF23 on AC are inconsistent, and further studies are needed to clarify the potential role of FGF23 in AC[35].
BSP is an acid glycoprotein synthesized by osteoclasts and osteoblasts and an essential component of MVs. BSP exists in blood vessels throughout the body and is an essential enzyme in forming calcium salts. It can hydrolyze phosphate bonds to increase native phosphate concentration and promote hydroxyapatite crystals formation[36].
1.5.2 Inhibiting factors
Currently, there are 6 types of CAC inhibitors:matrix γ-carboxyglutamic acid protein (MGP),pyrophosphate, fetuin-a, osteopontin (OPN), klotho and osteoprotegerin(OPG)[37].
MGP inhibits the differentiation of mesenchymal cells into osteoblasts by isolating the osteogenic and chondrogenic differentiation factor BMP-2, and the carboxylation of MGP is the main factor affecting its effect on BMP-2[38]. MGP carboxylation is performed by vitamin K-dependent carboxylase,so the inhibitory effect of the vitamin K antagonist warfarin on carboxylase will affect the inhibitory effect of MGP on CAC. Some studies have demonstrated that vitamin K antagonists promote CAC and valve calcification[39].
Fetuin-a, which is secreted by the liver and adipose tissue, combines with calcium and phosphate.Fetuin-a plays an inhibitory effect on CAC in VSMCs by preventing calcium phosphate growth and reducing calcium-induced apoptosis[40].
OPN has a two-way effect on the development of CAC. It can block the growth of calcified crystals by self-aggregation and adhesion of apatite crystals[41].OPN also inhibits calcification by stimulating the absorption of a mineralized matrix by osteoclasts.However, OPN also has a chemotactic effect on macrophages, produces pro-inflammatory properties,intensifies the inflammatory response, and enhances intimal calcification[42].
Klotho is related to anti-aging, which directly inhibits the phosphorus absorption of VSMCs. It is also essential for the activity of FGF23. FGF23 is synthesized by bone cells, which increase phosphate levels and reduce 25-hydroxy vitamin D to 1, 25-conversion of dihydroxy vitamin D. FGF23 increases the expression of OPG, an inhibitor of vascular calcification[43].
OPG is a phosphoprotein that regulates bone formation by inhibiting the growth of apatite crystals and osteoclast differentiation. It inhibits the maturation of osteoclasts by inhibiting the receptor activator of nuclear factor kappa B ligand(RANKL)[44-45].
In summary, the microcalcification of 5-500 µm formed in the early stage of CAC increases the destruction stress of atherosclerotic plaques and increases the risk of plaque rupture. With the progression and consolidation of calcification, the contact area between the calcified area and noncalcified area decreases, and the damage stress caused by microcalcification decreases. On the other hand,large calcified nodules increase plaque stability. Next,we would conclude the possible therapeutic effects of CHM from the pathogenesis in the early and middle stages of calcification.
2 Roles of CHM on CAC
2.1 CHM inhibites microcalcification
As part of the development of atherosclerosis,AC is caused by various factors, and it plays a role through multiple cellular, molecular signaling pathways. In the early stages of CAC, macrophage apoptosis, inflammatory cell necrosis, decreased expression of calcification inhibitors, and osteoblastlike cell differentiation is all essential factors that initiate the formation of arterial microcalcification.Therefore, theoretically, CHM with anti-apoptosis,anti-inflammatory, and anti-osteoblast differentiation effects can inhibit the occurrence and development of microcalcification. At present, most studies on the mechanism of CHM in the treatment of CAC focus on the anti-apoptotic pathway(Figure 2).
Radix salviae miltiorrhizae (RSM) is an extract from the rhizome of salvia miltiorrhiza. RSM has the effect of promoting blood circulation, removing blood stasis, and promoting new growth. It has been applied to the treatment of CAD, angina pectoris, myocardial infarction, arthritis for a long time. RSM contains a variety of active constituents, including hydrophile constituents, which include danshensu (DSS),salvianolic acid A (SAA), salvianolic acid B (SAB),and lipophile components, which include tanshinone I, tanshinone IIa, cryptotanshinone, and dihydrotanshinone.
DSS[46]inhibits ischemia-induced rat cardiomyocyte apoptosis and increases B cell lymphoma 2 (Bcl-2), and reduces the expression of BCL2-Associated X (Bax), through the activation of the mammalian target of rapamycin(mTOR),which is a crucial regulator of cell growth and proliferation.Paeonol and DSS combination has a significant cardioprotective effect on isoproterenol-induced myocardial infarction rats, by activating the nuclear factor erythroid-2-related factor 2/heme oxygenase 1(Nrf2/HO-1) signal and phosphoinositide-3-kinase/protein kinase B(PI3k/Akt) cell signaling pathway to inhibiting cardiomyocyte apoptosis[47]. DSS can also activate silent information regulator protein 1 (Sirt1)to inhibit excessive reactive oxygen species (ROS)through forkhead box O1 (FoxO1)/Ras-related protein seven signaling pathway and protect the heart from ischemia-reperfusion (I/R) injury[48]. This DSS effect can reduce the activation of oxidized low-density lipoprotein (ox-LDL) on ALP and further reduce vascular calcification.
SAA reduces the production of ROS and malondialdehyde in cells, increases the antioxidant enzyme activity of catalase (CAT), superoxide dismutase (SOD), and glutathione peroxidase.Further, by up-regulating Bcl-2 and reducing Bax and caspase-3, the ethanol-induced apoptosis was significantly inhibited[49]. SAA also increased the expression of klotho protein in SD rats with renal I/R injury[50], which may help to inhibit CAC. In I/R rat nerve cells, SAA regulates the activation of the wingless-type MMTV integration site family member(Wnt)/β-catenin signaling pathway by Dickkopf-1(DKK1) through enhancing the expression of microRNA (miR)-449a and attenuates brain injury,inflammation, and apoptosis[51]. SAA may reduce CAC through anti-oxidation and anti-apoptosis.
SAB can reduce the expression of inflammatory proteins,including c-Jun N-terminal kinase(JNK)and nuclear transcription factor kappa-B (NF-κB) and TNF-α in endothelial cells (ECs) and pericytes culturedin vitroinduced by ox-LDL. So SAB can protect ECs and pericytes from oxidative stress and apoptosis[52]. After SAB treatment of diabetic rats,endothelial nitric oxide synthase (eNOS)phosphorylation was significantly restored, Bcl-2 protein was significantly up-regulated, Bax protein was significantly down-regulated[53], and vascular endothelial cell apoptosis was reduced. These antiapoptotic effects of SAB can prevent or reduce the occurrence and development of microcalcification.
Tanshinone IIa,the main lipophilic component of RSM, can reduce lipid and calcium deposition under the intima of rat blood vessels,thereby reducing CAC.Tanshinone IIa minimizes the production of superoxide anions and inhibits the production of ox-LDL[54]. Ox-LDL can stimulate ALP activity, and then cause MVs calcification by generating local phosphate ions. This step is the key to force the CAC factor. Tanshinone IIa significantly increased the expression of Bcl-2 in apolipoprotein E knockout(ApoE-/-) mice and decreased the levels of Bax and caspase-3, thus reduced the ox-LDL induced apoptosis in VSMCs[55]. Tanshinone IIa may prevent microcalcification through anti-oxidation and antiapoptotic effects.
Notoginsenoside R1 (NGR1) is the main active component of panax notoginseng. NGR1 may inhibit the apoptosis of human umbilical vascular endothelial cells (HUVECs) induced by ox-LDL down-regulating miR-132 and then up-regulating MGP. NGR1 attenuates activation of JNK and NF-κB pathways by ox-LDL caused[56]and inhibited the inflammation and apoptosis process in the AS process. NGR1 can also reduce ox-LDL-induced apoptosis by up-regulating the expression of miR-221-3p in HUVECs[57]. NGR1 decreased calcium nodules and ALP activity of human alveolar osteoblasts in the TNF-α induced inflammatory microenvironment through inhibiting the NF-κB pathway and activating the Wnt/β-catenin pathway[58]. NGR1 can inhibit microcalcification through anti-apoptosis.
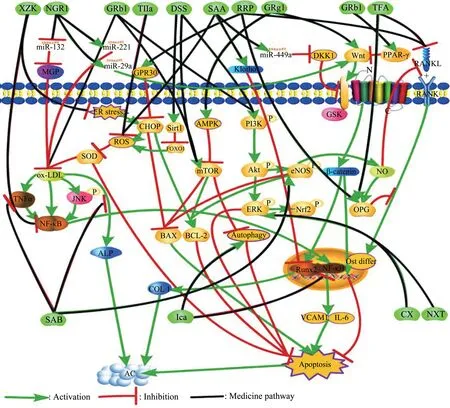
Fig.2 CHM inhibits microcalcification through the anti-apoptotic pathway
Ginseng is a commonly used CHM. It contains a variety of active ingredients and exerts its drug efficacy through multiple molecular pathways.Ginsenoside Rb1 (GRb1) is one of the active ingredients in ginseng. TongXinLuo is commonly used to treat CAD and angina pectoris, and its main component is GRb1. GRb1 reduces the apoptosis of HUVEC cells induced by TNF-α by inhibiting p38 and JNK signaling pathways. The protective effect of GRb1 can reduce the ratio of Bax/Bcl-2 and the level of caspase-3[59], further inhibiting cell apoptosis. In methylglyoxal-induced cell damage, GRb1 plays an anti-apoptotic effect by activating the PI3K/Akt signaling pathway[60]. GRb1 inhibits rat cardiomyocyte apoptosis induced by I/R injury through the activation of the mTOR signaling pathway[61-62]. GRb1 plays an anti-apoptotic effect through multiple molecular signaling pathways.These effects have prevented the formation of microcalcification to varying degrees.
Ginsenoside Rg1 (GRg1), another active component of ginseng, effectively induces autophagy in LDL receptor knockout (LDLr-/-)mice macrophages by activating adenosine 5'-monophosphate (AMP) -activated protein kinase(AMPK)/mTOR signaling pathway[63], further inhibited the apoptosis of macrophages. GRg1 also significantly reduces apoptosis induced by rat intestinal I/R injury by activating the Wnt/β-catenin pathway[64].
Rehmanniae radix preparata (RRP) is the main component of LiuWeiDiHuang (LWDH), which is used for ovarian extraction in ApoE-/-mice[65-66].LWDH inhibits the expression of CCAAT enhancerbinding protein homologous protein (CHOP) through G protein-coupled estrogen receptor 30 (GPR30) in the homocysteine(Hcy)treatment of HUVECs.In this study, LWDH reduced Hcy-induced apoptosis of HUVECs by regulating Bax/Bcl-2 and inhibiting the activation of caspase-3. RRP may also increase the expression of OPG and inhibit osteogenic differentiation through the RANKL pathway[67].
Red yeast rice is made by fermenting the fungus monascus purpureus on wet rice. It is the main component of XueZhiKang (XZK). It can inhibit cell apoptosis and increase the number of endothelial progenitor cells (EPCs) and reduce oxidative stress.The main component of RYR, named monacolin K, is the same substance as lovastatin[68]. XZK[69]treatment of ApoE-/-mice can significantly reduce endoplasmic reticulum stress (ER stress) in atherosclerotic lesions.ER stress plays a crucial role in the apoptosis of late,damaged macrophages, mainly mediated by the specific pro-apoptotic protein CHOP under ER stress conditions. XZK reduces the ratio of Bax/Bcl-2 by inhibiting the NF-κB pathway and further inhibits the mitochondrial apoptosis pathway of kidney cells in diabetic rats[70].
The main ingredients of Buchan NaoXinTong(NXT)[71]are astragalus, red peony root, RSM,angelica, and chuanxiong. NXT inhibits apoptosisrelated proteins through activated rat cardiomyocyte extracellular signal-regulated kinases (ERK1/2). NXT inhibits the expression of matrix metalloproteinase-2 and TNF-α mRNA in ApoE-/-mice[72].It increases the smooth muscle cell and collagen content of the plaque’s fibrous cap and reduces the accumulation and mineralization of macrophages in atherosclerotic lesions. Therefore, the combination of NXT and atorvastatin can stabilize plaque more effectively.
Astragalus is a commonly used CHM. The total flavonoids of astragalus (TFA) are the main component of astragalus. It can significantly inhibit the production of TNF-α and RANKL in rat serum and NF-κB pathway in synovial tissues of rats and promote serum OPG and OPG/RANKL ratio[73].These results indicate that TFA may inhibit CAC by regulating the OPG/RANKL/NF-κB pathway.
2.2 CHM promotes microcalcification
In the middle stage of CAC, microcalcifications further fuse to form larger calcified nodules.Although the fusion of microcalcifications reduces the junction area of the calcification area, the middle-stage calcified nodules still have the risk of causing plaque rupture.Therefore, in the middle stage of CAC, drugs that can promote the growth and fusion of microcalcification are needed to further fuse smaller calcified nodules into more extensive and higher density macrocalcification. In this way, the junction area of the calcified site can be reduced, the damage stress in the plaque can be diminished, and the risk of rupture of the plaque can be reduced. The promoting effect of statins on CAC may be the key reason for such drugs to reduce MACE events. For a long time,CHM has been used in the treatment of atherosclerotic diseases and has achieved good results. In addition to the anti-apoptotic and anti-oxidant effects of CHM mentioned above, it may also be related to the stabilizing effect of CHM on atherosclerotic plaques.The stabilizing effect of CHM on the atherosclerotic plaque is probably its stabilizing effect on arterial intimal calcification(Figure 3).
Among the various effects of RSM, the occurrence and development of microcalcification can be inhibited through anti-apoptosis and anti-oxidation.Still,domestic studies have also found RSM improves bone quantity and quality by favoring Wnt/β-catenin and OPG/RANKL/cathepsin K signaling pathways in ovariectomized rats[74]and promotes osteoblast differentiation and mineralization. Another study found that SAB promoted the ALP activity of human periodontal ligament cells by activating the Wnt/β-catenin signaling pathway, increased the expression of osteogenic differentiation markers, and increased mineralized nodules[75].These studies prove that RSM can inhibit not only the initiation of microcalcification in the early stage of calcification but also promote the stability of calcification fusion in the middle stage of calcification.
In addition to the anti-apoptotic effects mentioned earlier, NGR1 is also a kind of phytoestrogen that activates estrogen receptor (ER).NGR1 increases ALP activity and osteoblast gene transcription and produces type I collagen (COL1),osteogenic and runt-related protein 2 (Runx-2), a transcription factor that regulates osteogenic related genes in osteoblasts. NGR1 promotes the mineralization process of osteoblasts[76].
When GRb1 is incubated with rat mesenchymal stem cells (MSCs), it can increase the ALP activity expression and promote osteogenic differentiation[77]and may encourage the fusion of CAC calcified nodules. However, there are also reports that GRb1 inhibits the phenotypic transition of VSMC in CKD rats[78], and GRb1 treatment inhibited the Wnt/β-catenin pathway by activating peroxisome proliferator-activated receptor-γ (PPAR-γ) to reduce calcium deposition, ALP activity, and calcium concentration.Therefore, the molecular mechanism of GRb1 on arterial intimal calcification needs further verification.
Ginsenoside Rg3 (GRg3)[79], another component of ginseng, promotes bone formation in SD rats with al-induced osteoporosis by increasing the expression of calcium, phosphorus, serum ALP activity, COL I(collagen I), and osteocalcin. GRg3 also increases BMP-2 and promotes bone formation. The molecular mechanism of these effects of GRg3 and whether it can encourage calcified nodules'fusion is unclear.
In the same study[67], RRP was used to treat ovariectomized rats, which promoted bone formation by down-regulating DKK1 to activate the Wnt/glycogen synthase kinase 3 beta (GSK-3β)/β-catenin signaling pathway.These experiments show that RRP,like other CHM, plays a role in stabilizing atherosclerotic plaques through multiple signaling pathways.
Another study found that TFA stimulates BMP-2 and Runx2 proteins in SD rats[80]. It is essential for regulating the expression of bone matrix proteins such as osteocalcin, COL I, and osteopontin, and it is conducive to the formation of calcified nodules.Therefore, the mechanism of action of TFA needs further study. Recent studies have found that TFA can reduce the expression of FGF23 and Klotho by regulating the vitamin D (VD)-FGF23-Klotho pathway and improve the osteogenic ability of bone marrow MSCs culturedin vitro[81]. These TFA effects can also promote the growth and integration of microcalcifications, which is conducive to the stability of calcified plaques.
Icariin, as a medicine commonly used to treat fractures and osteoporosis, is the main component of many traditional Chinese medicine prescriptions.Icariin can increase the blood flow of cardiovascular and cerebrovascular and promote hematopoietic function and bone metabolism. Related studies have found that icariin induces osteogenic differentiation of bone marrow MSCsviaactivation of the Wnt/β-catenin signaling pathway[82]. The expression and translocation of β-catenin are required for osteoblasts to complete the differentiation process and promote mineralization. By increasing the expression and translocation of β-catenin, icariin can enhance the presentation of ALP,COL I,and osteocalcin(OCN)in osteoblasts and promote calcification. Icariin promotes Runx2 protein and gene expression by activating the Wnt/β-catenin pathway and ultimately promotes bone formation in mice[83]. Recent studies have found that icariin enhances the BMP2/Runx2 signaling pathway by up-regulating the expression of brain and muscle gene ARNT like 1 (BMAL1)[84], a biological clock gene, to induce osteogenic differentiation of bone marrow MSCsin vivo.Whether the calcification-promoting effect of icariin can promote the calcification of atherosclerotic plaques and whether it can promote the plaque lesions’ stability is a question worthy of further exploration.
Genistein is a kind of phytoestrogens. Someone used RNA sequencing (RNA-seq) analysis to observe the gene pathways that genistein affects the differentiation of mouse osteoblasts and found two upregulated genes (EregandEfcab2) that enhance osteoblast differentiation[85]. In comparison, 3 downregulated genes (Hrc,Gli, andIftm5) inhibit osteoblast differentiation. This difference in gene level was also verified in another study[86]. Genistein promotes the growth of rat osteoblasts and enhances ALP activity, and increases matrix mineralization. But for the primary medium arterial in this study, the VSMCs cultured with genisteinin vitrodid not promote osteoblast transformation. This result suggests that genistein may exert different therapeutic effects on bones and blood vessels through different genetic pathways, which will inspire expanding research ideas.
An aqueous fraction of Huogu (HGA) could enhance the mRNA and protein expression of Runx2,ALP, BMP2, OCN, and OSX.At the same time, HGA significantly increased the expression and secretion of β-catenin in a dose-dependent manner and nuclear translocation. Therefore, HGA promotes the osteogenesis of bone marrow MSCs through BMP and Wnt signaling pathways[87]. Gastrodin is a natural biologically active compound isolated from gastrodia.It is often used to treat cardiovascular and cerebrovascular diseases. Inin vitroexperiments,gastrodin increased the mRNA expression levels of osteogenic genesRunx2,OSX,BMP2,andosteocalcinin dexamethasone-induced osteoporosis mice[88].Ligusticum chuanxiong[89]also activates the BMP2/SMAD signaling pathway, which can up-regulate the expression ofRunx2gene and increase the osteogenic activity of human mesenchymal cells.

Fig.3 CHM promotes calcification fusion molecular pathway
These experimental studies have proved from different angles that CHM can promote the maturation and fusion of arterial calcification through other mechanisms of action and provide a theoretical basis for the treatment of CHM to stabilize atherosclerotic plaque and reduce cardiovascular events. However,the molecular pathways and clinical effects of different CHM components improving CAC still need more basic research and clinical trials to verify.
With the further development of calcified nodules, the contact area between calcified and noncalcified areas in the plaque decreased significantly,and the damage stress in the plaque decreased. Thus,in the later stages of CAC, calcified plaques change from vulnerable to stable plaques, reducing the incidence of cardiovascular events.
3 Conclusion
CAC is a type of calcification of the vascular intima, similar to the formation of intramembranous bone. It is a complication of atherosclerotic inflammation. The CAC mechanism is complicated and a multi-factor adjustment process. Apoptosis,inflammation, decreased expression of calcification inhibitors, and osteoblast-like differentiation is the main factors that initiate CAC.At present, there is no clear stage of CAC, but some studies have found that plaque calcification has different effects on atherosclerotic plaques at different stages of development. Therefore, in this review, according to calcification diameter, CAC was roughly divided into microcalcification of less than 5µm,microcalcification of 5-500 µm and calcified nodules of greater than 500 µm. Early microcalcification of AC with diameters ranging from 5-500 µm increases the destructive stress in atherosclerotic plaques. This microcalcification is an important cause of plaque rupture and cardiovascular events, as are the large necrotic core and the thin fibrous cap. However, with the development and fusion of arterial calcification,the contact area of the calcified area decreases, the destruction stress in the plaque decreases, and the atherosclerotic plaque tends to be stable.Therefore, in theory,at different stages of CAC,treatment strategies are different.
CHM is used as a traditional treatment for atherosclerotic diseases because of its effectiveness.Because CHM has complex components and has multi-target and multi-channel pharmacological effects, CHM has a different impact on various CAC development stages (Table 1). In the early stage of CAC, some elements of CHM reduce the occurrence of microcalcification by inhibiting apoptosis,inflammation, and osteoblast-like differentiation, and further reduce the plaque rupture caused by microcalcification. In the middle stage of CAC, some CHM elements promote plaque fusion and maturation by promoting osteoblast-like differentiation and promoting calcification. These effects can reduce the junction area of the calcified site in the atherosclerotic plaque, reduce the damage stress in the plaque, and stabilize the plaque lesion. The same CHM component may not only have an anti-apoptotic effect in the early stage of CAC but also play a role in promoting the fusion of calcified regions in the middle stage of CAC. This multi-channel pharmacological action may be an essential reason for the effective treatment of atherosclerotic diseases by CHM components.

Table 1 Mechanisms of CHM on AC

Continued to Table 1
In summary,CHM is used to treat atherosclerotic diseases, and it can reduce the vulnerability of atherosclerotic plaques to reduce cardiovascular events in many ways.This is also the theoretical basis for the effectiveness of CHM in the treatment of atherosclerotic diseases. More basic studies are needed to improve the molecular pathway of CHM in the treatment of CAC, and more clinical trials are needed to verify the effectiveness of CHM in the treatment of atherosclerotic diseases, to provide a more theoretical basis for CHM in the treatment of coronary artery disease.
