
表观遗传修饰调控阿尔茨海默病的研究进展*
2022-04-24 06:15林苏扬潘召韬马宇涛高君妍单江晖储超扬王清娟李丽萍
生物化学与生物物理进展 2022年4期
林苏扬 潘召韬 马宇涛 高君妍 单江晖 储超扬 谢 凯 沈 巍 王清娟 李丽萍**
(1)宁波大学医学院生理与药理学科,宁波 315211;2)宁波大学医学院附属医院康复科,宁波 315211)
阿尔茨海默病(Alzheimer’s disease,AD)是一种以进行性认知功能障碍和记忆减退为主要特征的神经退行性疾病,已成为威胁中老年人身心健康的主要疾病之一。AD 以细胞外β 淀粉样蛋白(amyloid β,Aβ)异常沉积形成老年斑、细胞内Tau 蛋白过度磷酸化形成神经原纤维缠结(neurofibrillary tangle,NFT)及神经元丢失等为主要的病理特征,临床表现为记忆障碍、失语、失用、失认、视空间技能损害、执行功能障碍以及人格和行为改变等。《2019 年世界老年痴呆症报告》统计显示,全球每3 秒钟就有1 例痴呆患者确诊,其中2/3 痴呆患者将发展成AD,到2050 年全球患AD人数将从目前4 700万上升至1.31亿[1]。目前,中国平均每年有30 万新发病例。随着人口老龄化日益严重,AD已成为全球最重要的社会医学问题之一。AD是一种由遗传因素(载脂蛋白E4、淀粉样前体蛋白、早老素1、早老素2 等基因突变)和非遗传因素(年龄、吸烟、嗜酒等因素刺激)相互作用引起的复杂疾病,但其发病机制尚不清楚,也尚无治疗AD的有效手段。携带相似或相同易感基因的个体在发生AD风险、其病理程度和临床表现中出现差异,提示表观遗传修饰可能在AD的异质性中发挥作用[2]。
表观遗传修饰是指在不改变DNA 序列的情况下,受环境因素影响导致基因表达发生可遗传改变。表观遗传修饰主要包括DNA 甲基化、组蛋白甲基化、组蛋白乙酰化、RNA 修饰和非编码RNA等,这些表观遗传修饰相互作用,可以调节基因表达,进而影响突触的可塑性、记忆的获得和巩固、神经通路的连接以及神经信号的传递等。大量研究表明,表观遗传学在AD发生发展过程中发挥重要的作用[3]。本文综述了近几年表观遗传修饰在AD发生发展中的调控机制,以期为表观遗传修饰作为AD治疗靶点或预测标志物的可行性提供新思路。
1 DNA修饰对AD的调控作用
DNA 甲基化是目前研究最广泛的表观修饰方式之一。为了探究DNA甲基化对AD发病的影响,West 等[4]证明,AD 患者颞叶APP 基因启动子甲基化水平低于非痴呆人群和非AD 型变性痴呆患者。随后,在AD 患者的额中回(middle frontal gyrus,MFG)和颞中回(middle temporal gyrus,MTG)中发现,5-甲基胞嘧啶(5-methylcytosine,5mC)和5羟甲基胞嘧啶(5-hydroxymethylcytosine,5hmC)总体水平显著提升,而在海马和内嗅皮层中均出现差异甲基化现象[5-7]。本文检索近几年DNA 甲基化修饰改变与AD发病风险相关程度的临床实验数据(表1),检测发现某一基因中5mC 或5hmC 修饰水平改变与AD发生发展存在相关性,提示该基因DNA修饰水平可以作为预测或诊断AD 病理程度。此外,在AD模型小鼠中采用生活环境刺激、药物诱导、过表达Tet催化区等治疗手段,发现通过改变DNA甲基化修饰水平可改善AD病理程度,提示通过干预DNA 修饰水平可以减轻AD 病理程度和提高记忆(表2)。
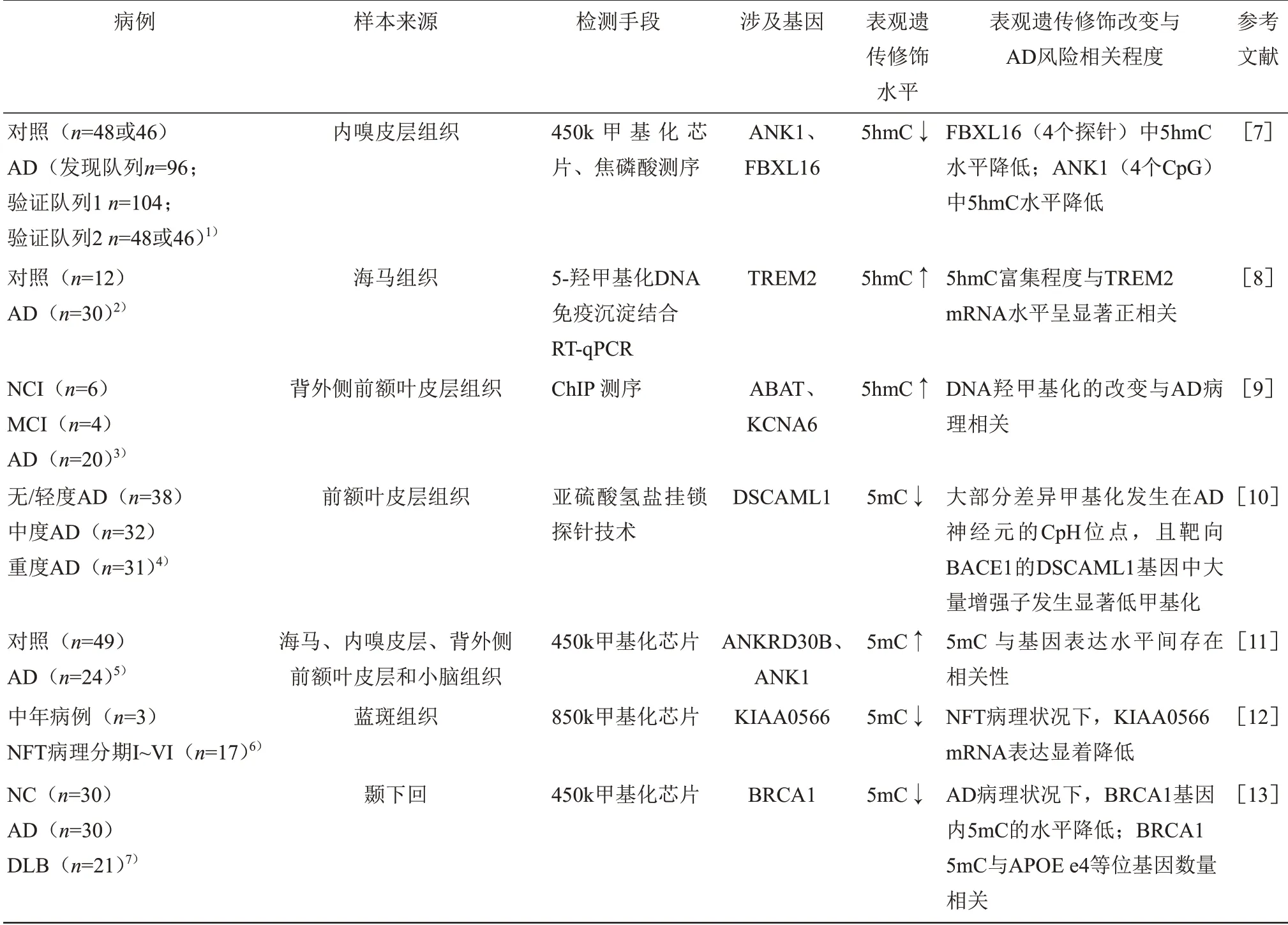
Table 1 Study on the relationship between genomic DNA methylation and AD risk in patient表1 患者基因组DNA修饰与AD风险相关性研究
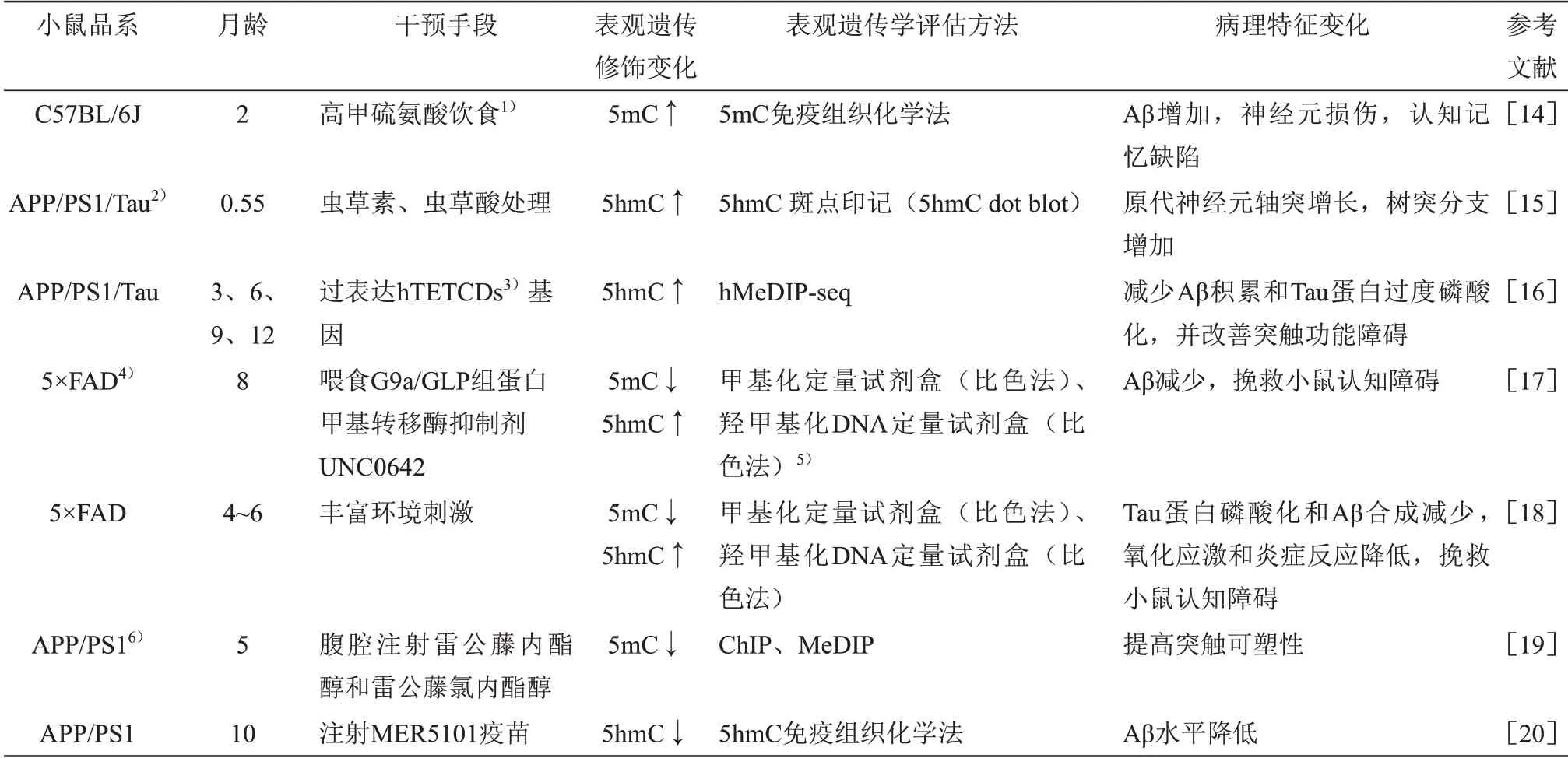
Table 2 Effects of different interventions mediated DNA modification on different strains of mice表2 不同干预手段介导DNA修饰对不同品系小鼠的影响
DNA 甲基化和去甲基化在神经发育和维持大脑突触可塑性过程中起重要作用。DNA 甲基化是指在DNA 甲基转移酶(DNA methyltransferase,DNMT)的作用下,由S-腺苷甲硫氨酸(S-adenosyl methionine,SAM)提供一个甲基基团,与DNA序列上特定的碱基共价结合的化学修饰过程。DNA甲基化生成物质主要包括5mC、N4-甲基胞嘧啶(N4-methylcytosine,4mC)和N6-甲基腺嘌呤(N6-methyladenine,6mA)。在哺乳动物中,目前研究比较多的是发生于胞嘧啶(C)-磷酸(p)-鸟嘌呤(G)二核苷酸(CpG)上胞嘧啶环第5 号碳原子上甲基化修饰(5mC)。C和G含量大于50%且序列长度大于200 bp 的CpG 聚集区称为CpG 岛。CpG岛主要位于基因启动子区域/转录起始位点的5'非翻译区(5'-untranslated regions,5'-UTRs),该区域中的甲基化可以抑制靶基因的转录,因此被认为是转录调控的形式之一[21]。其主要作用机制是:a.在CpG 岛基因启动子区域发生DNA 甲基化,抑制转录因子与该基因的启动子区域结合;b.甲基-CpG 结 合 蛋 白(methyl-CpG-binding proteins,MBDPs)与甲基化的DNA序列结合,从而发挥阻抑作用,抑制基因表达。目前研究表明,哺乳动物随着年龄的增长,血液中基因组整体DNA 甲基化呈现降低趋势[22]。DNA 去甲基化5hmC 修饰是一种不同于5mC 的表观遗传修饰。DNA 去甲基化主要通过Tet 蛋白家族催化5mC 转变为5hmC,5hmC 还可以进一步氧化形成5 甲酰基胞嘧啶(5-formylcytosine, 5fC) 和5 羧 基 胞 嘧 啶(5-carboxylcytosine,5caC),进而产生未甲基化的胞嘧啶C。另外,在胞嘧啶脱氨酶的作用下5hmC 也可以转成5 羟甲基尿嘧啶(5-hydroxymethyluracil,5hmU)。因此,5hmC也是一种DNA去甲基化的中间代谢产物。相比于5mC,5hmC在基因组总体水平较低且富含于中枢神经系统。5hmC通常富集于基因的转录起始位点(transcription start site,TSS)和5'-UTR 处,表明5hmC 与基因转录或翻译有关[23]。研究显示,5hmC在TSS上游区域、基因体和多聚腺苷酸化位点(polyadenylation site,PAS)下游区域中修饰水平与基因表达呈正相关,通过调控转录机制或作用于阻遏物来促进基因表达,从而影响大脑中枢神经系统的正常功能[24]。5hmC在正常神经发育和衰老过程中保持动态平衡,且其修饰水平的改变在一定程度上与AD 病理变化相关。5hmC水平在APP/PSEN1双转基因小鼠海马中随年龄老化而下降[25]。目前,DNA甲基化和去甲基化的改变与AD 病理之间相关性受到越来越多的关注。
AD 患者脑组织基因组5mC 改变影响AD 病理进程。5mC免疫组织化学分析AD患者脑组织病理切片,发现背外侧前额叶、小脑、内嗅皮层和海马中的DNA甲基化水平降低[11]。有研究指出,大脑中DNA 甲基化水平降低引起星形胶质细胞和小胶质细胞的活化,从而导致许多病理过程的恶性循环[26]。AD患者脑组织DNA甲基化与剪切Aβ的分泌酶表达存在相关性。AD的主要病因学假说是淀粉样变途径,该假说指出具有神经毒性的Aβ 寡聚体是由β 分泌酶和γ 分泌酶切割淀粉样前体蛋白(amyloid precursor protein,APP)后形成的。其中,β 分泌酶1(beta-site APP cleaving enzyme 1,BACE1)是大脑中β分泌酶的主要表达形式。体内实验证实喂食BACE1抑制剂NB-360后,APP转基因小鼠脑组织和脑脊液(cerebrospinal fluid,CSF)中Aβ 水平显著降低[27],表明抑制BACE1 可降低Aβ 表达水平。全基因组检测AD 患者前额皮层(prefrontal cortex,PFC)DNA增强子甲基化水平,发现BACE1 增强子低甲基化,提示在AD 中BACE1 低甲基化水平可能提高BACE1 mRNA 水平,进而促进Aβ 表达[10]。γ 分泌酶是膜内裂解蛋白复合物,负责裂解APP 膜内位点,并最终释放出Aβ。早老素1(presenilin 1,PSEN1)是γ 分泌酶的催化亚单位,通过调节γ分泌酶活性进而影响Aβ 生成[28]。研究发现在晚发性AD(late-onset AD,LOAD)大脑中PSEN1 低甲基化水平与AD进展程度相关,提示PSEN1 甲基化水平也可能通过 调 节Aβ 生 成 进 而 影 响AD 进 程[29]。此 外,PSEN1 突变导致AD 患者大脑区域微管相关蛋白Tau 启动子甲基化减少,提示PSEN1 也可调节Tau表达[30]。另外研究发现,AD 患者血清基因组DNA 甲基化的改变使其体内代谢产物水平发生异常变化,可能与Aβ 神经毒性及认知障碍存在关联[31]。血同型半胱氨酸(homocysteine,HCY)水平的增加、血清叶酸和维生素B12水平的降低被认为是引起AD发病的危险因素。机体内的一碳代谢包括叶酸和甲硫氨酸循环,正常甲硫氨酸饮食不能满足体内甲基化反应需求,机体需要摄入叶酸以提供甲基,而食物中的叶酸主要形式为5甲基四氢叶酸(5-methyltetrahydrofolate,5-Me-THF),在维生素B12作用下,5-Me-THE和HCY反应产生可以被细胞利用的四氢叶酸(tetrahydrofolate,THFA)和甲硫氨酸。因此,叶酸和维生素B12 是SAM 产生必不可少的元素。SAM 带有一个活化的甲基,参与甲基转移反应,通过充当主要的甲基供体,维持机体正常DNA 甲基化。在正常条件下,SAM提供甲基, 产生S- 腺苷同型半胱氨酸(S-adenosylhomocysteine,SAH),SAH 经过水解生成HCY,此后与甲硫氨酸相作用发生甲基转移,将HCY转变为SAM,该代谢循环需要不断供应叶酸和维生素B12。SAH 是DNMT 抑制剂,对诱导体内低甲基化水平有实质性影响[32]。体内研究表明,与喂食SAM组相比,喂食缺乏叶酸/B12/B6食物的APP 转基因小鼠血液表现出SAH 水平增加及PSEN1 启动子低甲基化,并引起PSEN1 和BACE mRNA 水平上升,最终导致神经元内Aβ 沉积和认知缺陷[33]。若在此基础上施用外源性SAM,可发现PSEN1 和BACE 甲基化水平下降,并降低了Aβ沉积[34]。因此,通过外源性给药提升SAM 水平,促进BACE1 的DNA 甲基化,可以缓解Aβ 产生和认知缺陷。此外,Aβ 斑块沉积能够引起降解酶如中性内肽酶(neprilysin,NEP)高甲基化,并下调NEP 表达,而NEP 低表达又促进Aβ 进一步沉积。NEP 是一种Aβ 降解酶,通过酶切清除Aβ 沉积。NEP在AD患者的脑内表达下降[35],而在AD小鼠中过表达NEP 能够显著降低Aβ 水平[36]。Aβ 通过使NEP 的启动子区域CpG岛的胞嘧啶高度甲基化,进一步抑制NEP 基因表达;而NEP 表达的减少则引起Aβ沉积。增多的Aβ又使NEP基因高甲基化,从而形成Aβ沉积和NEP高甲基化之间负反馈调节的恶性循环。
在哺乳动物机体内调控DNA去甲基化的Tet蛋白有3 个家族成员:Tet1、Tet2 和Tet3,它们的表达具有细胞类型特异性,在不同的细胞中调节不同的基因表达。在背角神经元中Tet1 通过提高DNA去甲基化水平而促进脑源性神经营养因子(brainderived neurotrophic factor,BDNF)的表达,进而增加突触可塑性[37];此外,研究发现,成年小鼠脑中Tet1 基因缺陷可使甘丙肽(galanin,GAL)、红蛋白(neuroglobin,Ngb)、含钾通道四聚化结构域14(potassium channel tetramerization domaincontaining 14,KCTD14)等神经发生相关基因启动子高甲基化,进而抑制基因转录,导致神经干细胞(neural stem cells,NSCs)自我更新能力及增殖分化能力降低,并进一步导致神经发生受损,提示Tet1介导DNA去甲基化水平调控神经元和NSCs功能而参与认知过程[38]。全身敲除Tet2 基因的小鼠,5hmC 水平降低,并抑制成体神经干细胞(adult neural stem cells,aNSCs)分化能力,提示Tet2 介导5hmC 调控NSCs 分化能力[39]。在体外细胞水平,过表达Tet2提高5hmC水平,可以促进老年2×Tg-AD 小鼠海马aNSCs 神经再生,提示Tet2介导5hmC 可能具有对抗AD 神经再生障碍的作用[40]。此外,在早期2×Tg-AD 小鼠海马区敲低Tet2,导致5hmC 水平降低,并引发早期AD 表现出晚期AD 相关的病理学特征,包括Aβ 沉积、GFAP 阳性星形胶质细胞增生、Iba1 阳性小胶质细胞过度生长以及促炎因子过度产生[40];在体内,老年2×Tg-AD小鼠海马区过表达Tet2,导致5hmC水平提高,并提高神经再生能力及减轻Aβ的负担,提示Tet2 是改善AD 神经再生障碍的靶标[41]。在神经前体细胞(neural precursor cells,NPCs)中敲除Tet3 导致全基因组5mC 水平降低、大规模的DNA低甲基化及Oct4、Nanog、Tcl1等多能性基因表达受抑制,提示在NPCs 中Tet3 通过调节DNA去甲基化而维持NSCs 多能性[42]。此外,研究发现,Tet3 通过JAK2/STAT3 信号通路调节NSCs 分化[43]。JAK2/STAT3信号轴是介导神经保护活动的主要传感器,激活JAK2/STAT3 轴可以抑制Aβ 相关的神经毒性,而失活JAK2/STAT3轴引起胆碱能神经元功能障碍,从而导致与AD 相关的记忆障碍[44]。目前,仍缺乏以Tet 蛋白为靶点治疗AD 的药物,但从动物实验研究表明,提高Tet蛋白表达可以增强神经干细胞增殖分化,促进神经再生,并改善学习和记忆障碍。
此外,目前已被证实DNA 去甲基化过程中伴随活性物质甲醛(formaldehyde,FA)生成[45]。正常生理大脑中的FA 水平较低,其含量约为0.2~0.4 mmol/L[46]。据报道与正常健康者相比,AD患者尿液和海马组织中FA 水平较高,提示高水平的FA 与AD 患者严重的认知缺陷可能存在关联[47]。已有研究证实,外源性FA过量接触或内源性FA代谢障碍均可导致FA 异常累积,是诱发与年龄相关认知障碍的原因之一[48]。将正常小鼠暴露于高浓度FA,发现小鼠出现认知缺陷且其大脑皮层发生Aβ 异常积聚、Tau 蛋白过度磷酸化等一系列早期AD样变化[49]。另一项动物体内实验证明,向恒河猴脑室内注射FA 导致其海马、内嗅皮层和前额叶皮层出现Aβ 沉积,同时加剧Tau 磷酸化,进而引起认知障碍[50]。目前认为,过量FA能够与Aβ42的Lys28 残基相互作用从而加速Aβ 聚集,此外Aβ 可抑制甲醛脱氢酶(formaldehyde dehydrogenase,FDH)的活性,从而减少FA 降解,导致FA 积累[51],因此形成过量FA与Aβ之间的正反馈循环。向APP/PS1 小鼠腹腔注射甲醛清除剂NaHSO3或辅酶Q10,导致AD小鼠大脑区域FA浓度降低,同时减少Aβ 聚集,改善APP/PS1 小鼠认知能力,从而缓解AD 病理症状[51]。因此,过量外源性或内源性FA可能是诱导AD发生发展的危险因素之一。
综上,DNA甲基化和去甲基化的过程影响Aβ生成、Tau 蛋白磷酸化和神经再生,引起AD 病理改变。SAH、PSEN1 和BACE 等基因的DNA 低甲基化修饰导致其表达水平升高,从而导致Aβ沉积。而Aβ 沉积使与Aβ 降解相关的基因如NEP 发生高甲基化,进一步加重Aβ沉积。DNA去甲基化过程中伴随FA 生成,而FA 异常累积,引起Aβ 沉积、Tau蛋白磷酸化,从而诱发AD。同时,Tet蛋白家族通过调控不同类型细胞中DNA 的去甲基化,如Tet1 调控神经元和NSCs、Tet2 调控aNSCs、Tet3调控NPCs等而影响神经再生。因此,提高机体叶酸、维生素B12含量以及过表达NEP,有利于减少Aβ 斑块生成,同时提高Tet 蛋白家族表达,保持5hmC 修饰高表达,有利于神经干细胞增殖分化,可以对抗AD神经再生和认知功能障碍。
2 组蛋白修饰对AD的调控作用
组蛋白是一种由H2A、H2B、H3和H4组成的八聚体,可与DNA交联形成核小体。组蛋白在其N端尾部发生修饰,影响染色体三维结构,最终导致基因转录改变。常见组蛋白修饰包括乙酰化、甲基化、磷酸化和泛素化[52]。本文总结了近几年组蛋白修饰与AD患者病变的关联性,研究发现组蛋白修饰水平改变参与AD 发病相关机制(表3)。在AD动物模型中,通过抑制剂或药物干预组蛋白修饰水平,可以对抗AD神经病理和认知衰退(表4)。

Table 3 Study on the relationship between genomic histone modification and AD risk in patient表3 患者基因组组蛋白修饰与AD风险相关性研究

Table 4 Effects of different interventions mediated histone modification on different strains of mice表4 不同干预手段介导组蛋白修饰对不同品系小鼠的影响
2.1 组蛋白甲基化/去甲基化与AD发生发展
已往研究认为,组蛋白中N-CH3键具有高热力学稳定性,因而难以去除甲基基团,甲基化修饰是不可逆转的。直到最新研究发现,组蛋白甲基化和组蛋白去甲基化在生命进程中处于动态平衡,组蛋白甲基化是在组蛋白甲基化转移酶(histone methyltransferase,HMT)催化下在氨基酸残基中添加甲基基团,而组蛋白去甲基化则通过组蛋白去甲基化酶(histone demethylase,HDM)将甲基基团脱去。
目前发现的HMT 主要有常染色质组蛋白赖氨酸甲基转移酶2 (euchromatic histone lysine methyltransferase 2,EHMT2/G9a)、混合谱系白血病 蛋 白1 (mixed lineage leukaemia protein-1,MLL1)、 SET 结 构 域 蛋 白1A (SET domain containing 1A,SETD1A)、SET 结构域蛋白1B(SET domain containing 1B,SETD1B)、ZESTE 同源 物 增 强 子2 (enhancer of zeste homolog 2,EZH2)、去甲基化酶有赖氨酸特异性去甲基化酶1(lysine-specific demethylase 1,LSD1) 以及含有Jumonji C 结构域(Jumonji C domain-containing,JMJD)蛋白家族。组蛋白甲基化的常见作用位点包括赖氨酸(Lys)残基上组蛋白H3 的K4、K9、K27、K36、K79 及 组 蛋 白H4 的K20,精 氨 酸(Arg)残基上组蛋白H3 的R2、R17、R26 及组蛋白H4 的R3,以及Lys 残基上组蛋白H1 的N 末端,这些氨基酸残基上不同程度的甲基化水平,大大增加了组蛋白修饰和基因表达的复杂性。组蛋白甲基化与转录激活和抑制有关,而且取决于其被修饰的氨基酸残基的位置及残基上甲基基团的数目。对CK-p25 AD小鼠海马组织进行ChIP-seq分析揭示了记忆认知功能的改变主要与位于启动子的H3K4me3 有关,而异染色质或多梳区域的H3K9me3 和H3K27me3 影响较小[65]。小鼠海马中H3K4me3和H3K9me2在长期记忆的形成过程中起重要作用。不同的是,H3K9me2与基因沉默有关,而H3K4me3 与基因激活有关[66-67]。G9a 是一种具有SET 结构域的组蛋白甲基化转移酶,参与体内H3K9me2 修饰[68],GLP 最初被描述为编码G9a 样蛋白的基因,与G9a在组蛋白上具有相同的底物特异性。G9a/GLP复合物与记忆和学习有关,参与长时程增强(long-term potentiation,LTP),维持突触可塑性以及上调BDNF[69]。用G9a/GLP 抑制剂UNC0642 作用于5×FAD 转基因小鼠,可以降低H3K9me2 并改变5mC 和5hmC 的总体水平,增加突触可塑性和减少神经炎症,防止Aβ 斑块积聚和提高认知能力[15]。因此,抑制G9a/GLP 活性可能是治疗AD 潜在的靶点。SET1/MLL 家族表达上调是导致机体内H3K4me3 异常升高的潜在原因,研究发现,抑制SET1/MLL HMT 催化活性,能够降低PFC 中H3K4me3 水平,恢复谷氨酸能突触受体表达且增加PFC突触可塑性,进一步改善AD小鼠认知障碍[63]。
目前已发现的HDM包括两种:赖氨酸特异性去甲基化酶(lysine-specific histone demethylase,LSD/KDM1),属于黄素腺嘌呤二核苷酸(flavin adenine dinucleotide,FAD)依赖性胺氧化酶的超家族;JMJD 蛋白家族,以Fe2+和α-酮戊二酸(αketoglutaric acid,α-KG)为辅离子,属于α-酮戊二酸氧化酶超家族,其中LSD1是第一个被发现的组蛋白去甲基化酶。在FAD的参与下,LSD1在体外可以特异去除H3K4的一甲基和二甲基修饰,在体内则可以去除H3K9 的一甲基和二甲基修饰。LSD1 参与NSCs 的正常表达,并参与学习和记忆的维持[70]。研究发现,敲除LSD1 能够激活Klf4、Myc 和Foxo1 3 个多能性基因,并诱发AD 病变,这表明LSD1 抑制多能性基因转录及阻止AD 发生[71]。同时,研究发现,在海马神经元中过表达LSD1 可以挽救细胞死亡,并抑制炎症反应发生,提示提高LSD1介导的组蛋白去甲基化有助于抑制细胞凋亡和炎症[72]。然而正常情况下LSD1分布在细胞核内,根据AD患者尸体检查发现,NFT中存在LSD1蛋白积累,且均异常定位于细胞质中,病理性Tau 蛋白可将LSD1 隔离在细胞质,并消耗细胞核中LSD1,从而加速神经元死亡[71-72]。含有Jumonji 域的蛋白质3(Jumonji domain-containing proteins,JMJD3)又称为赖氨酸特异性去甲基酶6B(lysine-specific demethylase 6B,KDM6B),是一种组蛋白H3K27 去甲基酶,在成人脑室下区域神经发生的重要激活剂。高表达JMJD3 将增强DLX2、MLL1和MASH1等相关基因H3K27me3去甲基化,进而促进神经元分化[73-74]。
综上,组蛋白甲基化和去甲基化与认知记忆能力有密切联系,通过调节G9a/GLP复合体和SET1/MLL HMT 家族活性,调节H3K4me3 和H3K9me2水平,影响认知相关基因表达;也可以通过作用LSD1 和JMJD3,调节神经元增殖分化,进一步改善AD认知障碍。
2.2 组蛋白乙酰化/去乙酰化与AD发生发展
组蛋白乙酰化和去乙酰化修饰过程分别由组蛋白乙酰转移酶(histone acetylases,HATs)和组蛋白去乙酰酶(histone deacetylases,HDACs)催化产生。HATs 使组蛋白发生乙酰化修饰,组蛋白乙酰化导致赖氨酸残基正电荷被中和,进而使染色质的正电荷与脱氧核糖核酸负电荷分离,核小体结构疏松,从而增强转录。HATs 包括CREB 结合蛋白(CREB binding protein, CBP)[75]、 p300[76]或p300/CBP 相 关 因 子(p300/CBP-associated factor,PCAF)[77],它们与短期记忆(short-term memory,STM)和长期记忆(long-term memory,LTM)维持有关。正常机体形成空间记忆过程中,海马区CBP、p300 和PCAF 表达和活性上调[77]。在海马区利用小干扰RNA (small interfering RNA,siRNA)或药物靶向抑制CBP 或p300 发现损害LTM,而抑制PCAF 会损害STM 和LTM,提示不同类型的HATs 对短长期记忆的作用效果有差异[78]。AD患者海马和额叶皮质均出现CBP表达水平的显著降低,且额叶皮质F2 区还发现PCAF 水平显著降低[54],AD患者颞叶以及APP/PS1小鼠海马中组蛋白乙酰化水平显著降低[58,79],提示HATs减少介导组蛋白乙酰化水平下降可能与AD记忆衰退有关。
HDACs 则通过去除Lys 残基末端乙酰基团而使组蛋白发生去乙酰化,从而抑制转录。AD患者脑区中高水平的去乙酰化酶与其认知记忆障碍相关。研究发现,AD患者海马和颞叶区域均可见高水平的HDACs[58,79],HDAC2 和HDAC6 在AD 患者的皮质和海马中过表达[80]。HDAC2可以通过调节PKA 依赖的环磷腺苷效应元件结合蛋白(cAMP-response element binding protein,CREB)磷酸化抑制记忆相关基因的表达[81]。在HDAC2过表达小鼠中,沿海马CA1 区锥体神经元和齿状回(dentate gyrus,DG)颗粒层神经元树突棘密度显著降低,且在CA1 区纹状体中突触素表达也显著降低,表明HDAC2 高表达则会抑制神经元结构和功 能[81]。因 此I 类HDAC,尤 其 是HDAC2 和HDAC3 被认为是记忆巩固的负调节剂[81-82]。与此相反,HDAC6 通过使微管蛋白去乙酰化,在改善微管稳定性中起关键作用[83]。HDAC 表达减少增加α 微管蛋白乙酰化,将促使泛素锌指结合域(binder of ubiquitin zinc finger domain,BUZ)与泛素化的底物蛋白结合,沿微管将异常蛋白运输到微管组织中心形成聚集体,从而经自噬-溶酶体通路清除错误折叠和聚集的蛋白质,通过进一步减轻异常蛋白质积累,发挥神经保护作用[84]。抑制HDAC6可有效清除Tau和Aβ[85]。随着不断深入研究, 发 现HDAC 抑 制 剂(HDAC inhibitors,HDACis)在对抗神经病理和认知障碍方面的具有潜在作用。Pan-HDAC抑制剂(如丙戊酸、曲古抑菌素A、4-苯基丁酸钠和伏立诺他(vorinostat))与锌依赖性HDAC蛋白(I类、II类和IV类)相互作用,参与调控Aβ 沉积及Tau 过度磷酸化,进而影响AD病理改变[86]。III类NAD+依赖性HDAC的竞争性抑制剂(如烟酰胺)选择性地抑制苏氨酸(Thr)第231 位点上Tau 磷酸化,并增加了乙酰化α 微管蛋白的表达,可用于治疗AD[87]。以上研究结果,提示HDACis在治疗AD具有潜在疗效。
综上,HATs 介导组蛋白乙酰化调控短期和长期记忆,提高HATs活性有助于记忆形成。HDACs介导的组蛋白去乙酰化参与神经元突触可塑性、Aβ沉积及Tau磷酸化,影响AD神经病理和记忆能力。HDAC2、HDAC3 和HDAC6 是记忆巩固的负调节剂。利用HDACis 抑制HDAC2、HDAC3 和HDAC6表达,可以清除Aβ和Tau蛋白,有效对抗神经病理和认知障碍。
3 RNA上m6A修饰对AD的调控作用
在mRNA上N6甲基腺苷(N6-methyladenosine,m6A)修饰水平的改变对神经干细胞分化及突触功能调控具有重要影响。目前已发现m6A 甲基转移酶有甲基转移酶3 (methyltransferase like 3,Mettl3)和甲基转移酶14(methyltransferase like 14,Mettl14),m6A去甲基酶有ALKBH5和脂肪量和肥胖相关蛋白(fat mass and obesity-associated protein,FTO),m6A结合蛋白有YTH 结构域家族蛋白(YTHDF1-3 和YTHDC1-2)。m6A 结合蛋白通过与mRNA 结合来提高翻译效率及加速RNA 降解。对处于增殖和分化条件下的aNSCs 样品进行MeRIP-seq显示m6A主要富集于神经发生和发育有关的转录本,因此m6A 水平异常可能影响神经发育相关蛋白表达,干扰正常神经功能[88]。
Mettl3 和Mettl14 作为m6A 甲基化转移酶,通过改变m6A 水平,调控神经分化程度及突触可塑性。研究发现,5×FAD 小鼠大脑中m6A 修饰水平相较于正常小鼠低[89]。已有研究证明,AD患者海马中Mettl3 表达降低[90]。在小鼠和人类胚胎干细胞中失活Mettl3 将导致m6A 水平降低,从而使神经元从自我更新到分化的过程受到严重抑制[91]。研究发现,在正常小鼠NSCs 敲除Mettl14 基因,m6A水平降低,导致NSCs增殖数量明显减少,并伴有过早的神经元分化,表明m6A 修饰能够促进NSC 自我更新并阻止细胞的过早分化,从而确保神经干细胞库的储备[92]。FTO 作为m6A 去甲基化酶,能够动态调控神经系统的发育及神经递质的传递,参与调控胰岛素,并诱发AD[93-94]。近期研究发现,在5×FAD 小鼠脑组织中,FTO 表达水平上调[89]。哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)是一种蛋白激酶,在胰岛素信号通路中起着至关重要的作用,与胰岛素缺陷导致的AD 有关[95]。在3×Tg-AD 小鼠脑组织中发现,FTO能够激活下游mTOR,促进Tau磷酸化[94]。相反,降低FTO表达将增加m6A水平,达到改善记忆的目的[96]。
另外,一些研究指出在m6A 修饰过程中,YTH 结构域家族蛋白能够增强突触信号传递效率及突触蛋白合成,从而改善小鼠学习记忆能力。敲除YTHDF1或YTHDF3发现神经元形态异常改变,自发兴奋性突触传递减弱并抑制突触蛋白的翻译[97]。利用shRNA 敲低小鼠Mettl3 或YTHDF1 发现海马突触信号传递和LTP受损,从而导致记忆能力及突触可塑性降低[98]。以上研究揭示,m6A 甲基化主要通过YTHDF1 和YTHDF3 促进记忆相关转录本的翻译,增加神经系统信号传递功能,从而提高学习记忆能力。另外,m6A 还可能在神经元修复和轴突再生中发挥关键作用。轴突损伤后发现m6A 水平升高,而YTHDF1 将促进参与轴突再生和恢复的蛋白质合成,导致mRNA翻译增强[99]。
综上,m6A 甲基转移酶Mettl3、Mettl14 和去甲基化酶FTO 协同作用调节神经元分化及突触可塑性,从而影响认知能力。m6A 结合蛋白YTHDF1 和YTHDF3 则在翻译水平上影响突触蛋白合成,从而改变神经系统信息传递效率,调控记忆发生。因此,通过靶向作用于Mettl3、Mettl14、FTO、YTHDF1 和YTHDF3 调控m6A 水平,可能有助于改善AD 中神经元凋亡和神经突触的丢失,进而改善记忆功能障碍。
4 非编码RNA对AD的调控作用
非编码RNA(non-coding RNA,ncRNA)指无法翻译蛋白质的RNA 分子。已有研究证实,在AD 患者外周循环(血清、血浆、外泌体、全血、外周血单核细胞)和脑脊液中检测到异常表达的ncRNA,其引起Aβ聚集、Tau磷酸化、神经炎症、突触可塑性和自噬等病理生理过程(表5)。通过调控ncRNA,策略有两种方式:a. 直接调节ncRNA 表达;b.应用siRNA 或ncRNA 模拟物操控ncRNA 表达。靶向抑制或促进ncRNA 表达,可影响下游BACE、APP 等靶基因的表达,进而治疗AD(表6)。因此,靶向调控ncRNA 可能成为AD的治疗策略之一,但仍需深入研究ncRNA 靶点以提高治疗效果。

Table 5 Study on the relationship between genomic ncRNAs and AD risk in patient表5 患者基因组ncRNAs与AD风险相关性研究

Table 6 Effects of different interventions mediated ncRNAs on different strains of mice表6 不同干预手段介导ncRNAs对不同品系小鼠的影响
4.1 miRNA与AD发生发展
microRNA(miRNA)是长度为22个核苷酸的小分子ncRNAs,广泛存在于血液、外泌体、CSF、脑组织等部位。目前已被证明是一种参与AD发生的潜在调节因子。
miRNA 最初以pri-miRNA 转录初产物形式存在于细胞核中,经RNase III 酶Drosha 加工转化为约由60 个氨基酸组成的茎环状miRNA 前体(premiRNA),随后通过输出蛋白5(exportin-5,Exp-5)和Ran-GTP 输出到细胞质中。在细胞质中,pre-miRNA 被内切核酸酶Dicer 进一步加工以产生双链miRNA。其中成熟miRNA同具有催化活性的Argonaute(AGO)蛋白一起形成RNA介导的沉默复合物(RNA-induced silencing complex,RISC),RISC 可通过结合特定信使RNA(message RNA,mRNA) 的3'非 翻 译 区(3'-untranslated regions,3'-UTRs),从而靶向抑制转录后翻译或诱导目标mRNA 及miRNA 降解,提示miRNA 通过结合mRNA 3'-UTR 来抑制目的基因表达。研究显示,AD患者病理改变同时伴随大量miRNA(miR-29c、miR-124、 miR-195、 miR-219、 miR-132/212、miR-101、miR-124、miR-193b等)表达水平下调,以 及 少 部 分miRNA (miR-7、miR-9、miR-34a、miR-125b、miR-146a 和miR-155 等)表达水平上调[113]。关于miRNA调控AD发病机制的研究非常广泛,但总体围绕5个方向研究:a.通过直接改变APP表达影响Aβ水平;b.通过改变BACE1表达影响Aβ水平;c.直接调节Tau磷酸化;d.参与突触可塑性;e.调控神经炎症发生发展。
miRNA可直接或间接调节Aβ形成。近年研究发现,miR-17-5p、miR-31-5p、miR-101-3p、miR-144-3p、 miR-153-3p、 miR-200c-3、 miR-381-3p、miR-383-5、miR-497-5p 均可以与人源或鼠源APP mRNA 3'-UTR相结合,降低APP表达[114-115]。用特定保护剂阻断miR-101-3p 与APP mRNA 3'UTR 相结合,导致HeLa 细胞中APP 表达水平增强[116]。在C57BL/6J 小鼠脑中敲除miR-101,抑制miRNA-101 与肝细胞核因子4α(hepatocyte nuclear factor 4α,HNF-4A) 结 合,使AMP 活 化 蛋 白 激 酶(AMP-activated protein kinase,AMPK)过度磷酸化,导致小鼠表现出认知功能障碍,提示miR-101介导HNF-4A/AMPK 通路参与维持认知功能[107]。与其他miRNA 不同的是,miR-346 与APP mRNA 5'UTR 相结合,能够上调HeLa 细胞APP水平,但在AGO 2表达减少情况下,miR-346活性降低,其促进Aβ 生成的能力减弱[117]。miR-29 家族(hsamiR-29a、hsa-miR-29b、hsa-miR-29c)特异性调节BACE1 间接影响Aβ 沉积。研究发现,AD 患者脑组织神经原纤维缠结周围发生TATA 结合蛋白(TATA-binding protein,TBP)异常积累,导致脑内干扰素γ(interferon-gamma,IFN-γ)释放增加,IFN-γ 激活STAT1 信号通路,从而抑制miR-29a/b表达[118]。在AD 患者脑组织和外周血中miR-29 表达显著下降,而BACE1表达和活性增强[119-120]。体外研究表明,在SH-SY5Y 细胞中提高miR-29c 水平,可以使得BACE1和APP-β蛋白水平显著降低,进一步分析发现miR-29c 通过靶向与BACE1 mRNA 3'-UTR 相 结 合,抑 制BACE1 表 达[120]。miR-29c-3p 还可以直接靶向与肿瘤坏死因子-α-诱导蛋白1(tumor necrosis factor-α-inducible protein-1,TNFAIP1) mRNA 3'-UTR 相 结 合,过 表 达miR-29c-3p 可以抑制TNFAIP1 表达,进而调节NF-κB 信号通路,减弱Aβ 沉积[121],提示miR-29通过抑制BACE1 和TNFAIP1 表达,降低Aβ 沉积。miR-31 能 够 降 低3×Tg-AD 小 鼠 海 马 中APP 和BACE1 mRNA 的表达,进而抑制海马中Aβ 沉积[114],表明miR-31 通过调控BACE1 表达间接影响Aβ沉积。
miRNA 可直接调节Tau 基因表达和代谢。miR-34a 在AD 患者脑组织和血液中表达上调,miR-34a 与Tau mRNA 3'UTR 结合,可以降低Tau蛋白水平[122]。miR-219在AD患者脑组织中表达下调,miR-219 表达减少促使Tau 蛋白合成,并加剧Tau 蛋 白 的 神 经 毒 性[103]。miR-128a 与BAG2 mRNA 3'UTR 结 合,降 低BAG2 表 达,而BAG2/Hsp70 复合物与微管相连,将Tau 蛋白输送至蛋白酶体进行降解,提示miR-128a 可以参与BAG2/Hsp70/Tau 降解机制[123]。蛋白激酶和磷酸酶之间的平衡决定Tau 蛋白磷酸化状态。miRNA 也可调控Tau 蛋白磷酸化。过表达miR-125b 可激活细胞周期蛋白依赖性激酶5(cyclin-dependent kinase 5,CDK5),进而诱导Tau 过度磷酸化[124]。miR-124通过激活非受体型蛋白磷酸酶1(non-receptor-type protein phosphatase 1,PTPN1)信号传导通路,可诱导糖原合酶激酶3(glycogen synthase kinase-3,GSK-3) 激 活 和 蛋 白 磷 酸 酶 2A (protein phosphatase 2A,PP2A)失活[125],提示miR-124参与调控Tau蛋白激酶/磷酸酶平衡。APP/PS1转基因小鼠海马和大脑皮层中miR-137 表达水平降低,钙电压门控通道亚基α-1C(calcium voltage-gated channel subunit alpha-1 C,CACNA1C)蛋白表达增加,提示miRNA 可通过调节电压门控钙通道介导钙进入神经元并调控如突触可塑性[126]。在rTg4510 小鼠(Tau 小鼠模型)大脑皮质神经元中过表达miR-142 出现胶质纤维酸性蛋白(glial fibrillary acidic protein,GFAP)和集落刺激因子1(colony stimulating factor 1,CSF1)的mRNA 水平增高,并伴随小胶质细胞和星形胶质细胞增生,提示过表达miR-142 可能诱发Tau 小鼠模型脑内神经炎症反应[127]。
综 上,大 部 分miRNA (miR-29、miR-31、miR-101、miR-346 等)直接或通过调控BACE1 间接影响Aβ 沉积,部分miRNA (miR-34a、miR-219、miR-128a、miR-125b、miR-124)间接调控Tau表达或直接影响Tau蛋白过度磷酸化,miR-137调控突触可塑性以及miR-142影响炎症发生,从而在多方面引起AD病理改变。
4.2 长链非编码RNA与AD发生发展
长链非编码RNA (long non-coding RNA,lncRNA)属于内源性ncRNAs,长度超过200 nt。研究发现,多种lncRNA 在脑组织中特异性表达,分布于细胞核、细胞质和线粒体中。lncRNA 参与调控Aβ 沉积、神经发育、突触可塑性和神经营养因子分泌等生物过程。迄今为止研究最多的lncRNA 是BACE1-AS,由定位于11 号染色体BACE1 基因座位(11q23.3)的对侧链转录而成,其作用是在mRNA 和蛋白质水平上调节BACE1 表达。BACE1-AS 可与BACE1 mRNA 形成双链结构复合物,避免BACE1 mRNA 被核酸酶或miR-485-5p降解,增加BACE1 mRNA稳定及蛋白质的高表达[128],提示上调BACE1-AS 可促使Aβ 生成。在AD 患者脑部持续产生的Aβ 可以诱导BACE1-AS表达上调,Aβ和BACE1-AS之间形成正反馈机制,进一步促进Aβ 表达,加剧病理恶化[129]。敲除BACE1-AS,可以改善AD 小鼠学习记忆能力[106]。通过判断血浆BACE1-AS 含量改变,可以诊断AD进展情况[130]。此外,BACE1-AS 还参与细胞损伤和细胞凋亡途径。自噬被认为是除降解酶外最重要的Aβ清除途径。因此,自噬-溶酶体系统调控异常被认为是病理条件下产生Aβ 的关键途径。自噬相关 基 因5 (autophagy-related genes 5,ATG5) 是AD 患者机体内一个关键的自噬基因,在AD 患者的血浆样本中表达增加[131]。BACE1-AS 通过与miR-214-3p 结合位点相互作用,导致miR-214-3p靶基因ATG5蛋白含量增加,从而促进自噬介导的神经元损伤作用[132]。黄连素治疗联合BACE1-AS敲除能够降低miR-132-3p表达,减轻Aβ25-35诱导的神经元损伤[133]。脑细胞质200(brain cytoplasmic 200,BC200)主要在海马和新皮质神经元的胞体和树突中表达[134]。BC200通过与真核起始因子4A(eukaryotic initiation factor 4,eIF4A)相结合,参与维持突触可塑性[135]。敲除BC200 能够抑制BACE1 表达,增加AD 细胞模型中细胞存活率[136]。但是,有研究指出AD 患者脑组织BC200表达增加,可能是由于神经元突触退化后发生代偿,导致BC200 发生错误定位及在体细胞的过度表达[137]。
参与神经营养因子分泌的lncRNA 主要有BDNF-AS和GDNFOS两种。BDNF-AS是BDNF的天然反义转录物,在机体内抑制BDNF表达。研究发现,敲低BDNF-AS 将提高BDNF mRNA 水平,增加BDNF 蛋白表达,有利于神经元生长和分化[138]。Aβ25-35诱导细胞内BDNF-AS 含量增加,导致嗜铬细胞瘤12(pheochromocytoma,PC12)细胞存活率下降。敲除BDNF-AS 后,PC12 细胞中BDNF 表达显著提升, 抑制细胞色素C(cytochrome C,CytC)释放,降低CC3(cleaved caspase-3)和Bcl-2 相关X 蛋白(Bcl-2-associated X protein,Bax)表达,提示BDNF-AS参与线粒体介导细胞凋亡[139]。以上结果表明,靶向抑制BDNF-AS 对治疗AD 有积极影响。胶质细胞源性神经营养因子(glial cell line-derived neurotrophic factor,GDNF)是一种重要的生物活性营养因子,能够营养神经细胞,促进神经元存活,并参与轴突损伤修复。机体内GDNF消耗与AD发病存在密切关系。lncRNA GDNFOS1和GDNFOS2是GDNF的反义转录物,可与GDNF mRNA 5'-UTR 外显子结合,抑制GDNF前体表达,导致GDNF蛋白水平下降[140]。而在AD患者血清中GDNF表达显著下调,提示lncRNA GDNFOS1和GDNFOS2可能参与调控GDNF表达,进而阻断神经保护作用[141]。
综上,随lncRNA 的研究不断拓展,发现lncRNA是AD的理想生物标志物。血浆BACE1-AS含量改变可诊断AD进展情况,同时BACE1-AS上调与Aβ 生成形成正反馈机制,加速机体细胞损伤和凋亡机制;脑组织过表达BC200,提高BACE1表 达 以 加 速Aβ 沉 积。 lncRNA BDNF-AS 和GDNFOS则分别通过抑制BDNF和GDNF表达,抑制神经发育。
5 结论与展望
表观遗传修饰在AD神经病理和认知功能中发挥重要的调控作用。临床分别采集AD 患者CSF、外周血或脑组织样本进行检测,发现异常的DNA修饰、组蛋白修饰、RNA修饰、ncRNAs等表观遗传修饰与AD发生发展密切相关,揭示了表观遗传修饰的改变与AD风险呈显著的相关性且可用于预测或诊断AD。表观遗传修饰是一种动态、可逆的变化过程,因此通过干预逆转异常的表观修饰,可达到改善AD 的目的。在AD 小鼠模型中,大量研究结果证明通过物理刺激(运动、丰富环境等)、药物(蛇床子素、当归芍药散等)、酶抑制剂(DNA 甲基转移酶抑制剂、HDAC 抑制剂等)、siRNA等干预方法调控酶的活性或水平,进而修正异常的表观修饰,可以调控Aβ 沉积、Tau 蛋白过度磷酸化、突触可塑性、神经炎症反应、神经营养因子释放等,从而抑制AD病理变化及改善小鼠认知能力(图1)。
由于临床实验数据采集的样本、病例数、疾病程度、检测手段等存在差异,导致部分实验结果相悖。目前临床使用操纵表观遗传修饰药物,主要是HDAC 抑 制 剂, 如 西 达 本 胺、 伏 立 诺 他(vorinostat)、罗米地辛(romidepsin)、帕比司他(panobinost)、贝利司他(belinostat)等,这些药物开发最初目的是抗癌,但是后来研究发现在治疗中枢退行性疾病中也发挥一定治疗作用。抑制剂治疗的瓶颈在于给药方式、药效持续时间和副作用。有些抑制剂药物因不能穿过血脑屏障,只能采用脑内植入和脑室内注射,这种创伤性的治疗方法给AD患者造成二次伤害;有些抑制剂药物半衰期很短,需要进一步研发脂质体、聚合物胶束或纳米粒为载体材料包裹;有些抑制剂药物会使得患者产生头痛、恶心、呕吐、腹泻甚至更为严重的副作用,且治疗成本高、疗效有限。应用药物、物理刺激或遗传学等手段干预调控各种修饰酶的表达水平,引起全基因组表观遗传修饰的广泛改变,但不能精确调控单基因的表观修饰,因此,目前尚缺乏特异性操控单基因表观遗传修饰的干预手段。研发以单基因、单一组蛋白或单一lncRNA或miRNA的表观遗传修饰为靶标的干预手段,更有助于理解表观遗传机制的具体作用过程。此外,在表观遗传机制中DNA修饰、组蛋白修饰、RNA修饰和ncRNA之间复杂的相互作用共同调控下游靶基因的表达,因此,未来研究需全面而系统的探究不同表观修饰在AD发生发展进程中内在关系。目前干预治疗措施大多针对AD 动物模型,在临床转化中对AD 患者的治疗效果仍需进一步探究。将来,重点研发有效的、安全的、低成本的操纵全基因组表观修饰的药物,或研发特异性操纵单个表观遗传学靶标的干预方法,全面而系统地探究表观遗传机制在AD病理中的调控作用,才能真正实现干预可行性、特异性和临床有效性三者合一的治疗效果。
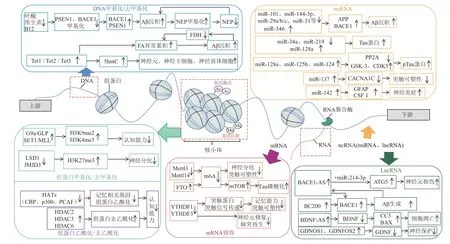
Fig.1 Mechanism of different epigenetic modifications regulate Alzheimer’s disease via multiple pathways图1 不同表观遗传修饰介导多种途径调控AD的作用机制
猜你喜欢
中国计划生育和妇产科(2022年5期)2022-11-16
上海师范大学学报·自然科学版(2022年3期)2022-07-11
中国农学通报(2022年13期)2022-05-31
电子产品世界(2021年8期)2021-01-16
健康之友(2020年1期)2020-03-24
福建基础教育研究(2019年10期)2019-05-28
中国计算机报(2019年49期)2019-02-07
中国新闻周刊(2017年36期)2017-10-21
创新时代(2016年8期)2016-10-21
