
典型聚乙烯、聚丙烯、聚苯乙烯废塑料共热解初期反应特性的ReaxFF分子模拟研究
2022-04-22 06:17:28贺兴处陈德珍
燃料化学学报 2022年3期
贺兴处 ,陈德珍
(同济大学 热能与环境工程研究所,上海多源固废协同处理和能源化工程技术研究中心,上海 200092)
城市生活垃圾中含有六种常见的塑料组分,包括高密度聚乙烯(HDPE)、低密度聚乙烯(LDPE)、聚丙烯(PP)、聚苯乙烯(PS)、聚氯乙烯(PVC)和聚对苯二甲酸乙二醇酯(PET)[1],其中,典型塑料组分PE/PP/PS是生产液态烃的原料。随着近年来城市生活垃圾的分类实施,塑料垃圾有条件单独收运或者废塑料可以容易地从干垃圾中分离获得。废塑料的回收利用方法主要分为物理和化学回收,由于物理回收技术的局限性,实际只有聚对苯二甲酸乙二醇酯(PET)和聚乙烯(PE)实现了物理回收和再循环,而且当塑料混合物的组成非常复杂时,再生塑料的使用性能会受到影响。化学回收是通过热解或化学分解等技术将废塑料中的有机成分转化成小分子烃等石油化工原料[2]。热解作为一种废塑料的化学回收技术,不仅可以解决废弃塑料对环境的污染,同时还可生产出具有极高经济价值的燃料油和其他化工原料[3],实现塑料垃圾的减量化、无害化和资源化利用,并且混合废塑料的热解是回收石化工业原料的一种经济有效的方法[4],并得到了广泛的关注。表1为PE/PP/PS的主要来源以及温度对热解产品分布的影响,可见低温热解主要以较大分子石蜡等产品为主,随着温度升高,产生更多的轻质油和气体小分子产品。
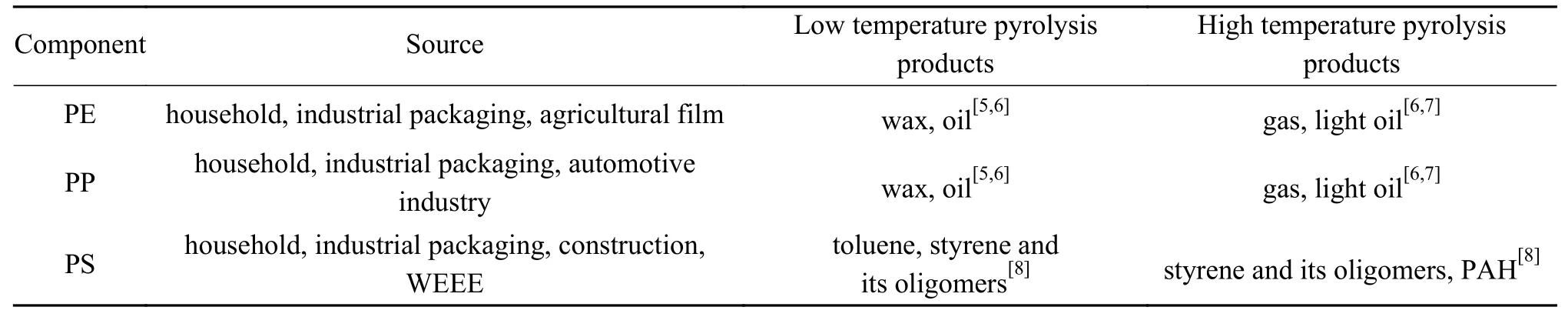
表 1 废塑料典型组分主要来源及其热解产品分布Table 1 Main sources of typical plastics and their pyrolysis products at low and high temperatures
Predel等[9]采用流化床反应器进行PE/PP/PS混合废塑料在510 ℃下的热解实验,通过Py-GC/MS等方法进行热解产物分析,结果表明,有价值的产物主要是从聚烯烃中得到的脂肪族蜡,同时也探究了不同混合比例在热解过程中的相互作用。Williams等[1]对混合塑料进行700 ℃下固定床热解研究,发现PE和PP热解所获得的油产品主要由烷烃、烯烃和烷二烯等脂肪族有机物组成,PS热解的油产品具有更多的芳香族成分,同时发现与PS混合的塑料比单一塑料热解具有更高的产气率和更低的产油率。Jin等[10]考虑到塑料的高黏度和低导热性,选用降膜热解反应器对PE/PP/PS在650 ℃下的快速共热解过程进行了研究,分析了三种组分在不同比例下的热解油气产率和热解产物组成,详细探讨了共热解特性。
近年来,随着反应分子动力学(Reactive Force Field Molecular Dynamics, ReaxFF MD)的迅速发展,基于原子层面的废塑料热解机理的研究也越来越深刻。ReaxFF MD是由 Van Duin 和Goddard共同发展的、将反应力场(ReaxFF)和分子动力学(MD)相结合、以描述复杂体系中分子键的断裂和生成的模拟算方法方法[11]。ReaxFF MD 已被广泛用于聚烯烃等的热分解过程的反应机理研究[12-14]。Liu等[12]将ReaxFF MD 应用于 HDPE 模型体系研究,通过分析模拟轨迹获取了PE热解的详细反应机理和气体分子生成途径,证明了应用 ReaxFF MD有助于建立对聚合物热解系统原子层面的机理理解。贺兴处等[14]采用ReaxFF MD 从原子层面详细揭示了CaO和H2O对PE热解过程的影响,揭示了CaO对碳链断裂的作用机理和H2O对产品品质的影响途径。但是目前关于ReaxFF MD探究PE/PP/PS混合热解机理的相关研究报道极少,而在实际工程中一般都是混合塑料共热解,要获得理想产品,研究其共热解机理非常重要。
鉴于PE/PP/PS在塑料垃圾中占比50%-70%的事实,本研究以PE/PP/PS为研究对象进行混合废塑料共热解机理研究,采用ReaxFF MD模拟手段,借助一款反应分子动力学模拟结果自动分析与可视化软件AutoRMA(automatic reaction mechanism analyzer)[15]探究原子层面共热解行为。AutoRMA基于LAMMPS[16]进行ReaxFF MD计算输出的键级文件和轨迹文件自动探测详细分子信息和化学反应信息,并以可视化的方式呈现分析结果。在用活化能验证模拟过程正确的基础上、分析不同混合比例和温度下热解产物分布,以了解混合塑料共热解与单一塑料热解的区别、及不同混合比例对热解产物的影响,便于产物的调节。
1 模拟方法
1.1 模型构建
模拟中首先使用Materials Studio(MS)[17]建立PE、PP、PS单链模型,然后利用Amorphous Cell模块构建周期性模拟盒子,体系密度设置为1.0 g/cm3,不同模拟体系添加分子的种类和数量如表2所示,其相对分子质量比例保持与Jin等[10]实验研究相同,综合考虑计算耗时与模拟精度,总原子数量控制在8000-10000。而后利用MS的Forcite模块对构建的模拟盒子进行几何构型优化和退火计算。
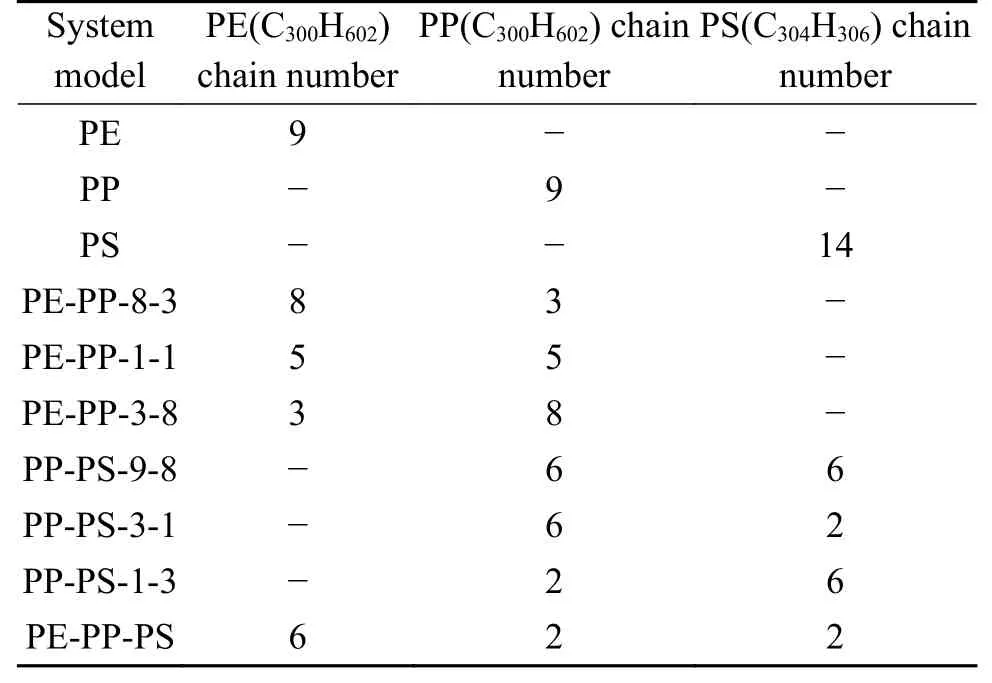
表 2 模拟体系分子构成Table 2 Molecular composition of simulation system
PE-PP-PS模拟盒子共有8436个原子(C3008H5428),图1为其在MS中进行几何优化和退火后计算后的构象。
1.2 模拟方法
利用LAMMPS的REAX包进行ReaxFF MD计算,所用力场参数[18]已用于聚合物的热解模拟[12,14,19]。当模拟体系较大时,ReaxFF MD极其耗时,已有研究[12,20-22]证明,在ReaxFF MD模拟中提高反应温度仅影响反应速率,对反应机制几乎无影响,因此,可以通过提高模拟温度以缩短反应所需的时间而不降低模拟精度[2]。因此,本研究进行了2400、2600、2800和3000 K一系列恒温模拟以研究共热解特性。所有计算采用NVT系综和周期性边界条件,时间步长为0.25 fs,cutoff为0.25。
2 结果与讨论
根据已有研究[23,24],热解产物按照碳原子数可分为炭(C41+)、油(C5-40)和可燃气(C0-4),油产物又可分为轻油(C5-13)和重油(C14-40)。由于各体系总原子数量不一致,因此,不同热解体系中可燃气分子的绝对数量缺乏对比性,因此,本文定义NR(Number ratio)为各气体分子数量与主要气体分子总数量的比值来进行对比,即:
2.1 共热解动力学分析
固相分解的单步动力学方程如式(2)所示,式(3)为反应速率的Arrhenius表达式,通过积分转换,可以将反应动力学函数f(α)表示为积分形式G(α), 进而在不同温度T下测定速率常数k(Ti),根据式(5)获得速率常数k(Ti)的Arrhenius图可以估计出活化能Ea和指前因子A。
式中,α为固体转化率,本研究中α的上限为0.9。R为理想气体常数。
由于废塑料热解反应主要包括碳链的断裂和重组以及自由基的解离和加成,C–C键(包括芳香键、单键、双键及三键)和C–H键的断裂对热解反应起着决定性作用,因此,可尝试C–C键和C–H键解离的动力学参数来表征整个热解体系的动力学参数。C–C键和C–H键断裂的反应速率常数k可通过式(6)求出[16,25]:
式中,N0为初始状态(t= 0)键数量,Nt为平衡状态(t=tequilibrium)键数量。进而利用Arrhenius方程式可求得活化能Ea和指前因子A。
整个体系活化能可通过体系中C–C键和C–H键的活化能加权求和进行计算,即为Bonds方法,在体系刚开始热解过程没有H–H键变化,因此,不进行H–H键相关计算。
采用RTP(reduced time plots)方法对体系在2400、2600、2800和3000 K下进行热解计算结果进行分析,可获得废塑料热解过程的动力学参数。图2为2400-3000 K下PE-PP-PS体系等温热解过程C–C键和C–H键的数量变化及动力学计算,其中,TG、Bonds和Char Bonds分别表示按照固体转换率α在0.9之前的失重速率、根据体系中炭油气三项产物中总化学键断裂速率和只根据体系炭产物中化学键断裂速率进行统计计算。
图2(c)为在不同温度下体系热解250 ps过程中的失重变化,随着热解过程进行,失重率迅速增大并趋于平衡,另外随着温度的升高,体系在热解后期会出现失重率降低的情况,这是由于在热解模拟计算过程中没有实时去除挥发分产生结焦现象,导致部分挥发分重新结合形成焦炭。如图2所示,随着温度升高,C–C键和C–H键的断裂速率逐渐增大。在相同温度下,Bonds(统计整个体系中的化学键)方法比Char Bonds(只统计炭产物中的化学键)方法计算具有更大的键断裂速率,说明在热解初期炭裂解为小分子的同时,该小分子会进一步裂解为更小分子,并且由于C–C键和C–H键的数量随时间具有一致的变化趋势,因此,小分子形成的主要方式是碳链的断裂和C–H生成H自由基和H2。根据图2中C–C键及C–H键数量变化可以发现,C–C键和C–H键断裂速率在16 ps之前较16 ps之后更大,形成较为明显的两个变化趋势,尤其是2800和3000 K温度条件下;另外,在16 ps左右,2800和3000 K下体系中C–C键和C–H键数量变化均达到一个3 ps左右的短暂平衡态,此时热解体系中C–C键和C–H键数量第一次趋于稳定, 称之为第一次达到平衡状态。由于模拟过程在高温下进行,之后产物的二次反应大量发生,所以此稳定状态非常短暂。由于本文研究目的是探讨PE/PP/PS共热解初期的相互作用机理,暂不考虑二次反应,因为二次反应与所处的反应条件有关,不适用于分析不同废塑料共热解的相互作用机制与动力学参数;为确保二次反应没有大量发生,并保证反应率α不大于0.9,选择C–C键和C–H键断裂速率一致的反应区域进行计算[26],可获得更能揭示PE/PP/PS共热解本质的动力学参数而不受二次反应的干扰,因此,选择tequilibrium=16 ps作为计算时间以保证所有计算温度下反应率α从开始到该时间的一致性。下一步再采用类似文献[27]的研究方法进行二次裂解和炭产品相关分析。采用Char Bonds方法计算结果如图2(f)所示,其与文献[10]实验结果基本一致,并且Bonds方法较TG方法更接近实验值,因此,热解过程C–C键和C–H键解离的动力学参数可以准确表征整个体系的动力学参数,故其余体系采用化学键数量变化方法进行动力学参数计算。
图3为不同温度下PE/PP/PS单独热解及以典型混合比共热解过程C–C键和C–H键的数量变化,其中,Char-Bonds-C-C和Char-Bonds-C-H是基于Char Bonds方法统计的C–C键和C–H键的数量变化,Bonds-C-C和Bonds-C-H是基于Bonds方法统计的C–C键和C–H键的数量变化。可见,随着温度升高,整个体系的C–C键和C–H键数量迅速降低,碳链的解离速度明显加快。在相同温度下,C–C键的减少程度大于C–H键,这是由于在热解初期,碳链断裂主导整个过程,·H自由基的脱离稍晚于碳链的断裂,炭产物中C–C键和C–H键的变化趋势一致也表明,·H自由基尚未大量脱离。
PE/PP/PS典型混合比例体系共热解的动力学参数计算结果如表3所示,其中,实验值来自Jin等[10]对废塑料典型组分在降膜热解反应器中的共热解特性试验研究,模拟时间取体系第一次达到平衡状态的时间16 ps,可见活化能的模拟值与实验值的误差仅为 ± 3.86%。
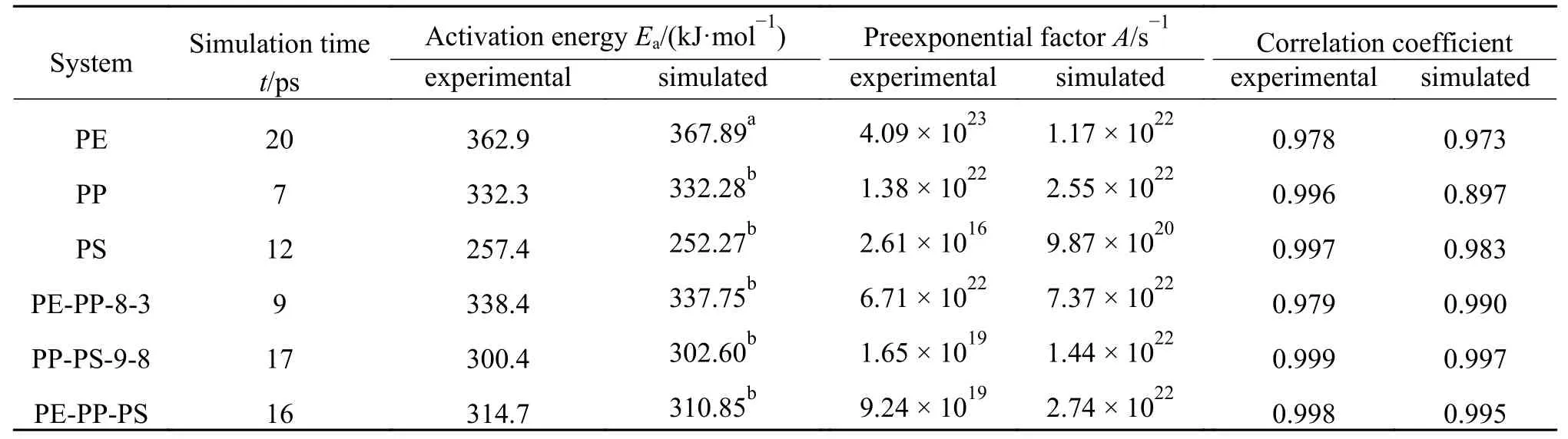
表 3 PE/PP/PS废塑料单独热解及共热解过程动力学参数Table 3 Pyrolysis kinetic parameters of PE, PP, PS and mixtures of PE-PP, PP-PS and PE-PP-PS
由表3可以看出,除PE体系外,采用Char Bonds方法可以得到与实验相符的活化能,而采用Char Bonds方法计算纯PE体系热解20 ps得到的活化能是532.63 kJ/mol,明显大于实际热解活化能,这是由于PE没有支链结构,在热解初期长链断裂的同时,热解产生的中间产物的分解对整个反应有较大影响,因此,采用Bonds方法进行计算更符合实际热解过程。
2.2 共热解特性分析
2.2.1 PE/PP共热解特性
图4为3000 K下PE/PP混合热解过程三相产物的产率变化,所有体系在20-40 ps均达到90%以上的转化率,混合热解的转化率在PE和PP单独热解平均转化率之间,产物油和气的质量比分别在60%和40%左右,与Williams等[1]和Jin等[10]实验结果相符。由于PP具有支链结构,PP热解开始时间较PE早,但其构成单体丙烯C3H6分子碳原子数大于PE构成单体乙烯C2H4分子,因此,在初始阶段PP的固体转换率变化低于PE。所有体系在20 ps左右,焦油产量达到最大,一次裂解基本完成,之后随着二次反应的进行,焦油分子进一步分裂,产生更多的气体分子。
在一次裂解主导阶段,随着PP掺量的增加,转换速率逐渐变缓,过多的PP掺量会导致期间焦油产量减少,而增加产物炭比率。但在二次反应开始后,随着PP比例的增加,体系中焦油产率逐渐增加,而气体产率逐渐减少,这也主要是由于构成单体碳原子数的影响。因此,以油为目标产物的热解中,增加PP/PE中PP的比例可以提高热解油的产率,而增加PP/PE中PE的比例可以提高不凝气体的产率。但在较短停留时间工况下,需要根据焦油性质确定合适的PP掺量。
图5为不同比例PE/PP在3000 K混合热解80 ps后油品中重油和轻油的相对比率,油产品中轻质油组分比例大于重油组分,少量PP的加入不影响油的组分,但随着PP掺量的增加,重油组分逐渐增多,而轻油组分减少。因此,以轻质油为产品的废塑料热解,PP掺量需要进行适当控制。
图6为不同比例PE/PP在3000 K下混合热解80 ps的主要气体分布,随着PP掺量的增加,H2和CH4产量呈上升趋势,这是由于PP具有支链结构,在热解过程更易脱落形成·CH3和·H自由基,形成类似PE无支链结构,因此,造成C2H4相对含量的下降和C2H6相对含量的相对平稳。
2.2.2 PP/PS共热解特性
与PE/PP共热解类似,如图7所示,PP/PS共热解油产率随热解时间的延长均呈现先上升后随着二次裂解发生出现下降趋势,并且掺有PS的体系中,由于PS热解过程推迟,热解到80 ps尚未达到完全平衡。在40-60 ps之后,体系热解炭的产率开始上升,这是因为在热解过程中,没有将挥发分分离,产生了结焦现象。由于本研究主要分析热解油和气产品,因此,取第一次平衡态60 ps的模拟结果进行产物分析即可,不影响分析结果。随着体系中PS掺量的增加,热解产品中炭和油的产率升高,而气体产率降低。
图8为不同比例PP/PS在3000 K下混合热解60 ps后油品中重油和轻油的相对比率,由于PS单体为苯乙烯C8H8,分子量较大,因此,PS参与的混合热解体系更倾向于产生更多的油,而且油产物中重油组分的比例也较高。随着PS掺量增加,油产物中重油组分逐渐增多,轻油组分产率逐渐减少,但在PP∶PS比例达到1∶1后其比率变化较小。
图9为不同比例PP/PS在3000 K下共热解80 ps后的主要气体分布,随着PS相掺量增加,H2含量呈上升趋势,这是由于混合热解中PS会大量产生·H自由基,而CH4呈下降趋势。另外适量的PS可以促进PP裂解为其单体C3H6,但是过量会呈现抑制作用。
2.2.3 PE/PP/PS共热解特性
分别取PE-PP-PS混合体系在不同温度下第一次达到平衡状态时的计算结果研究温度对热解的影响,2400、2600、2800和3000 K下的等温模拟第一次达到平衡状态的时间分别为250、120、95和45 ps,图10为不同温度下PE-PP-PS体系热解主要产物的产率及固体转化率,不同温度下转化率均能达到93%以上,温度为2400-2600 K,转化率迅速升高了4.78%,但是随着温度继续升高,转化率开始降低,这是由于在混合热解过程中发生了结焦。高温有利于重油裂解为轻油,随着温度的升高,重油和轻油的相对含量发生明显变化,轻油的相对含量从2400 K的44.77%升高到3000 K的56.18%。图11为PE-PP-PS体系在2400-3000 K不同温度下热解产品气主要组分,随着温度升高,H2和CH4的产率逐渐上升,但C2H4和C3H6的产率先上升后降低,这是由于高温会促使烃类小分子进一步裂解生成相对更小分子产物,但如前所述,过高的温度会造成结焦而不是进一步裂解,因此,促进聚合物大量分解为其单体结构需要选择适当的温度。
2.3 共热解机理分析
表4为PE/PP/PS混合模拟体系在典型比例、3000 K下化学反应进程追踪结果,两条反应进程分别表示碳链的断裂和塑料单体及小分子的生成时间顺序,其中,R–CH3表示不包含CH4的气体小分子。

表 4 3000 K下PE/PP/PS在等温混合热解化学反应进程Table 4 Chemical reaction process during isothermal co-pyrolysis of PE/PP/PS at 3000 K
碳链断裂顺序三个时间节点分别表示开始反应时间、碳链全部参与反应时间和转换率达到95%的反应时间,由于PS热解过程会产生结焦现象,无法达到95%以上的转换率,因此,只有前两个时间节点。如表4所示,相比单一塑料热解,共热解初始反应时间延迟,但总体反应时间缩短,说明不同塑料之前具有相互促进作用,能够加快反应速率,这与Li等[28]报道一致。并且产物更倾向于为较小分子,共热解加剧了反应物之间的相互作用。PE和PP单独热解时,首先生成其构成单体分子,继而生成烷烃和小分子气体,但混合热解过程呈现相反趋势:首先生成烷烃和小分子气体,而后生成其单体。PS热解首先产生苯乙烯和苯,继而生成小分子气体,但在混合热解体系中,苯的产生时间明显延迟,这是由于苯乙烯较稳定,而且PE和PP产生的含碳自由基倾向于和·H自由基结合,因此,PS在热解过程中更倾向于提供·H自由基,从而减弱了苯环和乙烯基之间碳碳键的断裂,如图9在PP/PS混合体系中,随着PS掺量的增加,乙烯的产量不增甚至有微弱下降,也是因此原因。
PE/PP/PS混合热解反应在2-4 ps开始产生H2和·H自由基,因此,表3中活化能的计算若只根据C–C键断裂速率进行计算会导致结果偏低,加权C–C键和C–H键活化能更符合实际热解过程,可以获得更准确的动力学参数。
3 结 论
本研究利用ReaxFF进行了废塑料典型组分PE/PP/PS共热解特性研究,利用AutoRMA进行产物演变分析,从动力学、热解产物及热解过程三方面揭示了原子层面的共热解特性。研究结论如下:
PE/PP/PS共热解的动力学参数可通过C–C键和C–H键断裂的活化能加权求和获得,与实验数据接近。
增加PP-PE体系中PP的含量可以提高油和可燃气的产率,增加PP-PS体系中PS的含量可以提高炭和油产率。
在PE-PP-PS体系中,高温利于重油裂解为轻油,同时高温也会促使烃类小分子进一步裂解生成更小分子,随热解温度升高,H2和CH4的产率明显上升,但C2H4和C3H6的产率先上升后降低。相比单独热解,混合热解开始反应时间有所延迟,但达到第一次平衡的总反应时间缩短,并且更倾向于生成较小分子产物。
PE和PP单独热解时,首先生成其单体,继而生成烷烃和小分子气体,但两者共热解过程中首先生成烷烃和小分子气体,而后生成其单体;另外在共热解中PS更倾向于提供·H自由基从而与PE和PP生成的自由基结合,形成小分子烷烃或H2。
上述研究结果为混合废塑料共热解产物的控制奠定基础。
猜你喜欢
云南化工(2021年10期)2021-12-21 07:33:22
云南化工(2020年11期)2021-01-14 00:50:48
应用化工(2020年9期)2020-09-29 08:55:16
Coco薇(2016年8期)2016-10-09 16:58:11
中国资源综合利用(2016年4期)2016-02-08 17:40:52
中国资源综合利用(2016年5期)2016-02-03 02:56:23
少儿科学周刊·少年版(2015年3期)2015-07-07 20:57:25
应用化工(2014年7期)2014-08-09 09:20:23
中南民族大学学报(自然科学版)(2014年4期)2014-08-06 05:49:24
声学技术(2014年2期)2014-06-21 06:59:06
