
长非编码RNA NEAT1在糖尿病及其并发症中的分子调控机制
2022-04-21 04:02袁凤英孙少康金智生
中国生物化学与分子生物学报 2022年2期
袁凤英, 孙少康, 金智生
(甘肃中医药大学, 兰州 730000)
糖尿病作为一种常见的内分泌系统疾病,其特征是由于胰岛素分泌不足或胰岛素抵抗而导致的持续性高血糖状态引起的一组代谢综合征[1]。根据国际糖尿病联合会最新的统计数据显示,2019年全球糖尿病患病约4.63亿人,预计到2030年会上升到5.78亿人[2]。目前,糖尿病病因及发病机制仍未完全阐明,且易引起眼、肾、心、血管、神经等多组织器官不可逆的严重损害,临床缺乏有效的治愈手段。因此,进一步探索驱动糖尿病发生发展的分子调控机制,鉴定出糖尿病及其并发症的特异性生物标志物和分子治疗靶标,无疑是阻止糖尿病发生发展,改善其患者生存质量的有效策略。
随着高通量测序技术及全基因组学的发展,越来越多的数据表明,长非编码RNA(long non-coding RNA, lncRNA)是机体正常生命活动以及疾病发生发展中重要的调控因子,其异常表达和突变是引起糖尿病、心脏病、癌症等许多疾病的主要原因之一[3-5]。lncRNAs的最早发现可追溯到上世纪90年代,以lncRNA的 X非活动特异性转录本(X inactive specific transcript,Xist)为代表[6]。之后,研究者将超过200个碱基的非编码RNA归类为lncRNA[7]。lncRNA是RNA聚合酶Ⅱ/Ⅲ(polymerase Ⅱ/Ⅲ,Pol Ⅱ/Ⅲ)在基因组中的任何缺乏开放阅读框区域的转录产物[8]。尽管lncRNA不编码任何蛋白质,但它可作为指导分子、诱饵分子、支架分子以及信号分子,与DNA、RNA、蛋白质相互作用发挥其生物学功能,进而影响人类许多疾病进程[9, 10]。
核旁丛组装转录本1(nuclear paraspeckle assembly transcript 1,NEAT1)是近年来新发现的lncRNA分子, 它在多种哺乳动物细胞中广泛表达。越来越多的研究表明,NEAT1在各种生物学和病理过程中起着重要调控作用。例如:NEAT1在胰岛β细胞中的表达上调,可作为信号分子激活信号转导和转录活化因子(signal transducer and activator of transcription 3,STAT3)相关炎症信号通路诱导细胞凋亡,进而介导小鼠血糖升高并产生胰岛素抵抗[11];在宫颈癌中,NEAT1可作为诱饵分子与miR-133a竞争性结合,进而诱导miR-133a的靶基因SRY盒转录因子4(SRY-box transcription factor 4,SOX4)表达上调,促进宫颈癌细胞增殖、侵袭及迁移[12];此外,NEAT1亦可作为miR-144-3p的分子海绵,调节NF-κB信号通路的活性介导心肌细胞的损伤[13]。鉴于lncRNA NEAT1在人类疾病中具有重要的调控作用以及丰富的生物学功能,本文对NEAT1在糖尿病发生发展中的作用进行归纳总结,并详细解读NEAT1在糖尿病及其并发症中的分子调控机制及其相关生物学功能,以期为糖尿病早期预防、诊断以及分子靶向治疗提供新的科学参考。
1 NEAT1简述
基于GeneCards在线数据库信息显示,NEAT1作为一种基因间lncRNA位于人类11号染色体q13.1区域(如Fig.1所示。数据来源于网站:https://www.genecards.org)。
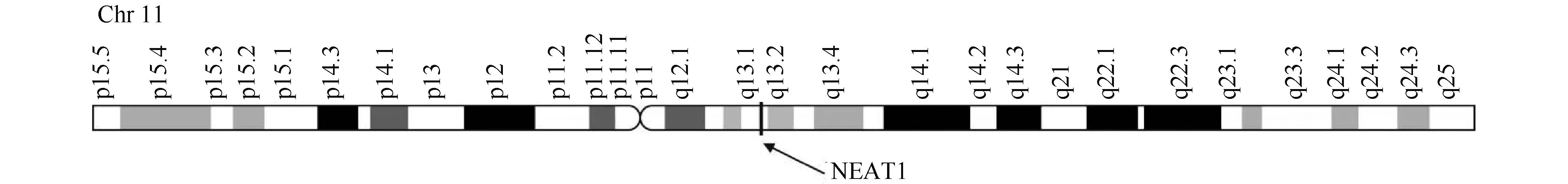
Fig.1 Localization of NEAT1 on human chromosome 11 (From GeneCards database, database website:https://www.genecards.org) The information collected in the online database shows that NEAT1, as an intergenic lncRNA, is located in the q13.1 region of human chromosome 11
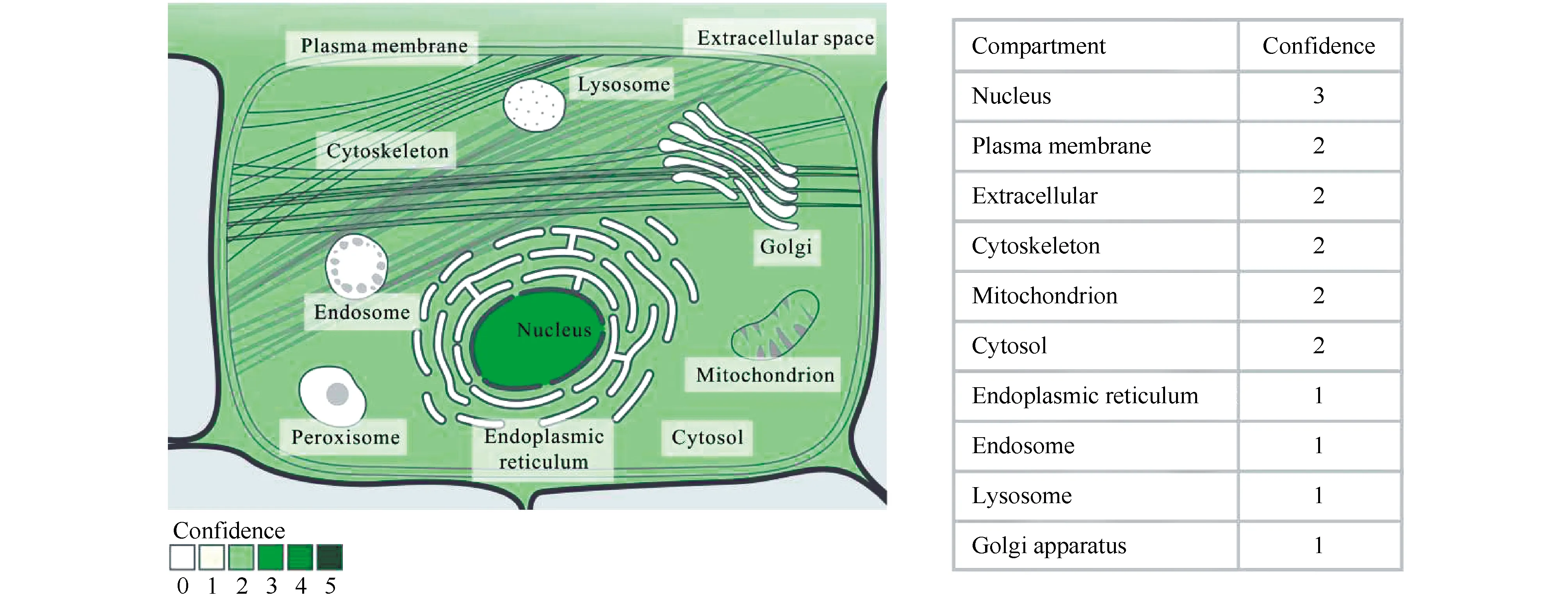
Fig.2 Subcellular localization of NEAT1 (From GeneCards database, database website: https://www.genecards.org) Subcellular localization showed that NEAT1 was expressed both inside and outside the cells, but mainly located in the nucleus
NEAT1在细胞内外均有表达,但主要定位于细胞核中(如Fig.2所示。数据来源于网站:www.genecards.org)。以往的研究表明,NEAT1在胰岛β细胞中的表达显著上调,是诱导血糖升高并产生胰岛素抵抗的重要原因[11]。表达上调的NEAT1可通过与微小RNA(microRNA,miRNA)竞争性结合、调控细胞信号传导通路等多种方式参与调节糖尿病心肌病、糖尿病肾病、糖尿病眼病进展,在糖尿病及其并发症中发挥着关键调控因子的作用[14-16]。更为重要的是,根据已有的临床样本数据分析表明,NEAT1在糖尿病患者中表达显著上调,并与体重减轻、伤口愈合缓慢等不良反应呈显著相关性,可能是临床治疗糖尿病的新分子靶标及糖尿病早期预防、诊断的有效生物标志物[17]。
2 NEAT1在糖尿病及其并发症中的分子调控机制
糖尿病是一类多种因素引起的高血糖代谢疾病,主要表现为胰岛素分泌不足、或(和)抵抗,进而累及机体多器官功能受损[18]。糖尿病的病因本质上是多因素的,例如:先天免疫因素、遗传因素、环境因素以及基础疾病的影响等。基因组学研究发现,lncRNA NEAT1的异常表达是引起糖尿病及其并发症的重要因素之一,是糖尿病及其并发症的重要调节因子。
2.1 NEAT1与糖尿病
在血糖升高的小鼠模型的胰岛β细胞中发现NEAT1的表达显著上调。高表达NEAT1通过激活STAT3相关炎症信号通路诱导细胞凋亡,进而引起血糖升高并产生胰岛素抵抗[11]。为了进一步探索lncRNA在糖尿病中的生物学功能,Lin等[19]通过构建1个与2型糖尿病相关的ceRNA网络,发现miR-181b与NEAT1、mTOR之间具有相互作用位点;分子机制上,NEAT1可作为miR-181b的分子海绵,诱导下游mTOR的表达上调,参与促进糖尿病进程。此外,在200名受试的2型糖尿病患者中,NEAT1的表达增加了5.28倍,且NEAT1高表达与患者体重减轻(P=0.04)、疲劳(P=0.01)、伤口愈合缓慢(P=0.002)、视力模糊(P=0.008)、食欲不振(P=0.007)等不良反应呈正相关[17]。
2.2 NEAT1与糖尿病并发症
2.2.1 NEAT1与糖尿病心肌病 糖尿病心肌病是糖尿病最常见的并发症,也是糖尿病相关死亡的主要原因[20]。越来越多的数据表明,高血糖介导的活性氧产生是糖尿病心肌病发生的主要诱导因素。过度的活性氧产生诱导氧化应激异常信号级联反应,触发心肌细胞凋亡,导致心脏纤维化和重塑[21, 22]。多项数据表明,NEAT1在引起糖尿病心肌损伤方面发挥着至关重要的调控作用。
NEAT1在糖尿病心肌细胞中表达显著上调。上调的NEAT1通过与miR-140-5p竞争性结合,提升miR-140-5p的靶基因HDAC4的表达水平,进而诱导心肌细胞凋亡[23]。在另一项研究中,NEAT1亦可作为miR-27b的分子海绵上调PINK1的表达,加重糖尿病心肌损伤[24]。值得注意的是,梓醇(中药材地黄的有效成分)能够减少心肌细胞内的活性氧的产生,降低NF-κB的转录活性,下调NEAT1的表达,抑制心肌细胞凋亡。机制分析表明,氧化应激促进了NF-κB的转录活性,入核NF-κB能够与NEAT1启动子结合并促进NEAT1的表达,诱导心肌细胞凋亡[25]。另外,在大鼠糖尿病模型中观察到,石斛合剂(一种用于治疗糖尿病的中药,由石斛、黄芪和丹参组成)与二甲双胍联合用药能够靶向NEAT1,使核转录因子E2相关因子2(nuclear factor erythroid 2 related factor 2,Nrf2)上调,有效改善心脏组织形态并减少心肌细胞凋亡[26]。
2.2.2 NEAT1与糖尿病肾病 糖尿病肾病是糖尿病最严重的微血管并发症之一,也是引起肾衰竭的主要原因[27]。糖尿病肾病的病理特点主要包括系膜细胞增殖、外基质积聚、纤维化以及足细胞减少[28]。由于糖尿病肾病的发病是多因素的,包括炎症、上皮间质化(epithelial-mesenchymal transition,EMT)、氧化应激、线粒体损伤、凋亡以及表观遗传的改变等,其潜在机制尚未完全阐明[29]。越来越多的数据表明,NEAT1通过与miRNA竞争结合、激活细胞异常信号传导通路等调控方式引起糖尿病肾病发生并使之加重。
在136例2型糖尿病肾病患者和25名健康受试者中发现,NEAT1在糖尿病肾病患者中的表达显著上调。多变量回归分析表明,NEAT1与糖尿病肾病诱导因子:突触素、肾损伤分子1(kidney injury molecule 1,KIM-1)、尿N-乙酰-β-D-葡萄糖苷酶(N-acetyl-beta-D-glucosamidase,NAG)、miR-21、miR-124呈正相关。与糖尿病肾病保护因子:表皮生长因子受体(epidermal growth factor receptor,EGFR)、miR-93、miR-29a呈负相关(P< 0.0001,R = 0.702)。肾小球肥大是糖尿病肾病早期形态学改变的主要特征之一[16]。Liao等[30]发现,高糖条件下系膜细胞中STAT3被激活从而诱导NEAT1转录,表达水平上调的NEAT1进而与miR-222-3p形成双链RNA,从而限制了miR-222-3p与细胞周期素依赖性激酶抑制剂1B(cyclin dependent kinase inhibitor 1B,CDKN1B)的结合,引起肾小球肥大。在糖尿病肾病的小鼠模型中,NEAT1的表达显著上调,敲除NEAT1后系膜细胞增殖减少、凋亡增加;并观察到纤维化标志物(纤维链接蛋白、Ⅰ型胶原)以及炎性因子(TNF-α、IL-1、IL-6)表达减少[31]。同样,在糖尿病大鼠及高糖诱导的小鼠系膜细胞中,NEAT1表达显著上调,并观察到Akt/mTOR信号的高度激活,进而使转化生长因子β1、纤连蛋白和Ⅳ型胶原的水平上调;数据表明,NETA1通过激活Akt/mTOR信号通路促进系膜细胞增殖和纤维化,从而加重糖尿病大鼠的肾损伤[32]。在另一项研究中,NEAT1被证明是miR-27b-3p的分子海绵,使miR-27b-3p靶基因ZEB1的表达上调,诱导肾纤维化形成和EMT[33]。此外,NEAT1亦可通过与miR-23c竞争性结合,诱导凋亡信号调节激酶1(apoptosis inducing signal regulated kinase 1,ASK1)、α-平滑肌动蛋白(α-smooth muscle actin,α-SMA)、波形蛋白以及纤维连接蛋白表达增加,促进系膜细胞纤维化和EMT[34]。Klotho是一种抗衰老蛋白质,在糖尿病肾病发展过程中对肾小管EMT起保护作用。新的证据表明,Klotho通过下调肾小管细胞中NEAT1的表达抑制ERK1/2信号传导,以防止EMT和肾纤维化,说明NEAT1可能是糖尿病肾病潜在的治疗靶点[35]。焦亡是一种程序性细胞死亡的炎症形式,由Nod样受体3 (nod-like receptor 3,NLRP3)炎症小体复合物形成后,胱天蛋白酶-1(caspase-1)激活介导,导致细胞裂解和促炎细胞因子释放。焦亡被认为是引起糖尿病肾病慢性炎症的重要因素。据报道,糖尿病肾病模型中,NEAT1的表达上调与焦亡发生有关。就分子机制而言,NEAT1通过与miR-34c竞争性结合,从而诱导miR-34c下游基因NLRP3、胱天蛋白酶1、IL-1的表达介导肾小球系膜细胞焦亡[36]。
2.2.3 NEAT1与糖尿病眼病 糖尿病眼病是糖尿病的常见微血管疾病并发症,也是糖尿病引发失明的主要原因[37]。越来越多的数据表明,血管内皮生长因子(vascular endothelial growth factor,VEGF)信号通路在糖尿病眼病中发挥关键作用[38]。炎症反应是机体对糖尿病的主要反应之一,当炎症部位的血管形成时,血管内皮生长因子的水平上升会伴随炎症反应加剧。尽管炎症反应在短期内对机体是有益的,但长时间的代谢压力和炎症会对视网膜造成永久性损伤,包括水肿、免疫细胞入侵和疤痕等[39]。在高糖诱导的人视网膜内皮细胞损伤模型中NEAT1表达显著上调,当NEAT1被敲除时,VEGF以及炎性因子(Cox-2、IL-6、TNF-α)的水平随之下降,有效减缓了糖尿病视网膜病变的发展[40]。视网膜色素上皮细胞EMT被认为是糖尿病视网膜病变发生的另一重要因素。最新报道指出,NEAT1在高糖条件下的视网膜色素上皮细胞中表达上调,它可作为miR-204的分子海绵,介导下游基因SOX4的表达上调,启动EMT进程[41]。糖尿病白内障是糖尿病早期眼部并发症,也是致盲的另一主要原因。据报道,在糖尿病白内障患者的晶状体中,观察到阴阳1(Yin-yang 1,YY1)(一种重要的锌指结构蛋白转录因子)显著减少。YY1可通过直接与NEAT1启动子区域结合来调节NEAT1的表达。当YY1低表达时,NEAT1的表达随之下调,下调的NEAT1与miRNA-205-3p竞争性结合减少,进而介导miRNA-205-3p下游靶基因基质金属蛋白酶16(matrix metalloproteinase-16,MMP16)的表达被下调,从而影响糖尿病白内障进程[42]。
3 问题与展望
由于糖尿病的发病机制尚未完全明确,目前临床治疗主要以控制临床症状、延缓其并发症的发生发展为主,尚无治愈方法。探索糖尿病及其并发症的发病机制,鉴定出具有特异性的糖尿病的生物标志物和分子治疗靶点迫在眉睫。研究发现,NEAT1作为一种lncRNA分子,是引起糖尿病及其并发症发生发展的重要因素。从机制上讲,表达上调的NEAT1可通过与miRNA竞争性结合、调节细胞信号传导通路等多种方式参与调节糖尿病、糖尿病心肌病、糖尿病肾病与糖尿病眼病进展。更为重要的是,目前的临床资料显示,NEAT1在糖尿病及糖尿病肾病患者中的表达显著上调,与伤口愈合缓慢、疲劳等多种不良反应呈显著相关性。总之,对lncRNA NEAT1在糖尿病及其并发症的分子调控机制进行梳理分析表明,它可能是糖尿病早期预防、诊断及分子靶向治疗的有效生物标志物,具有极其重要的临床应用价值。但目前关于lncRNA NEAT1在糖尿病及其并发症中的研究数据主要源于细胞及动物实验,需收集更多的临床数据,进一步阐明lncRNA NEAT1在糖尿病及其并发症中的分子调控机制,完善其在糖尿病中的系统调控网络,使其尽早服务于临床。
猜你喜欢
中国种业(2022年9期)2022-10-13
中国畜牧兽医(2022年9期)2022-09-22
中学生数理化·高一版(2021年12期)2021-09-05
家庭科学·新健康(2019年10期)2019-11-18
健康必读·下旬刊(2019年7期)2019-07-29
现代农业科技(2018年13期)2018-10-20
家庭医学·下半月(2017年7期)2017-08-08
大众健康(2017年3期)2017-04-13
中国中药杂志(2016年20期)2016-11-19
科学中国人(2016年9期)2016-11-04
