
芴基胺基二甲基钛配合物催化的乙烯和降冰片烯及其衍生物共聚研究
2022-04-20 03:54:44芦风正郑宇超孙延杰蔡正国
合成技术及应用 2022年1期
芦风正,郑宇超,孙延杰,蔡正国
(1. 东华大学材料科学与工程学院,上海 201620; 2. 东华大学纤维材料改性国家重点实验室,上海 201620)
基于环烯烃与α-烯烃的共聚产物(环烯烃共聚物,简称COC)是一种具有高透光度、高耐温性、低双折射率、高耐腐蚀性以及低吸水性等优异性能的无定型聚合物[1-2],多被用于光学、医疗和电子信息等领域[3-8]。对于我国而言,此类高性能、高附加值聚烯烃材料的市场需求正急剧增长,但相关生产与应用技术却长时间被国外所掌握,国内需求长期受国际环境制约,因此进行COC材料的生产技术开发以及为日后该产品的工业化生产与应用技术积累资料刻不容缓。镜头用COC材料需满足玻璃化转变温度(Tg)>135 ℃、透光率>90%、折射率>1.54以及熔体流动速率(MFR)>36等主要性能指标,这些性能指标可通过共聚过程中链结构调控来达到。
单茂过渡金属催化剂(如图1所示)具有较小的空间位阻从而使得环烯烃单体的反应活性大大提高,例如Shiono等[9]采用的催化剂体系甚至可以直接催化NB均聚,且即使在NB插入率较高的前提下仍能保持高的催化活性,这在其他类型过渡金属催化剂中是比较少见的。王华金等[10]通过改变芴环或胺基上的取代基合成了具有不同空间位阻和电子效应的芴基胺基二甲基钛配合物,并通过在0~20 ℃条件下利用MMAO作为助化剂催化NB单体与乙烯单体共聚反应。结果表明,胺基配体上的取代基显著增加了阳离子钛中心周围的空间位阻,较大的空间位阻使得NB单体插入速率降低。但有趣的是,虽然配合物空间位阻较大但其在较低温度0 ℃下也实现了高活性(高达1 620 kg/(mol·h))的NB与乙烯共聚,其值随着各催化体系中进料NB投料比的增加而降低,同时伴随着共聚物分子量的降低。
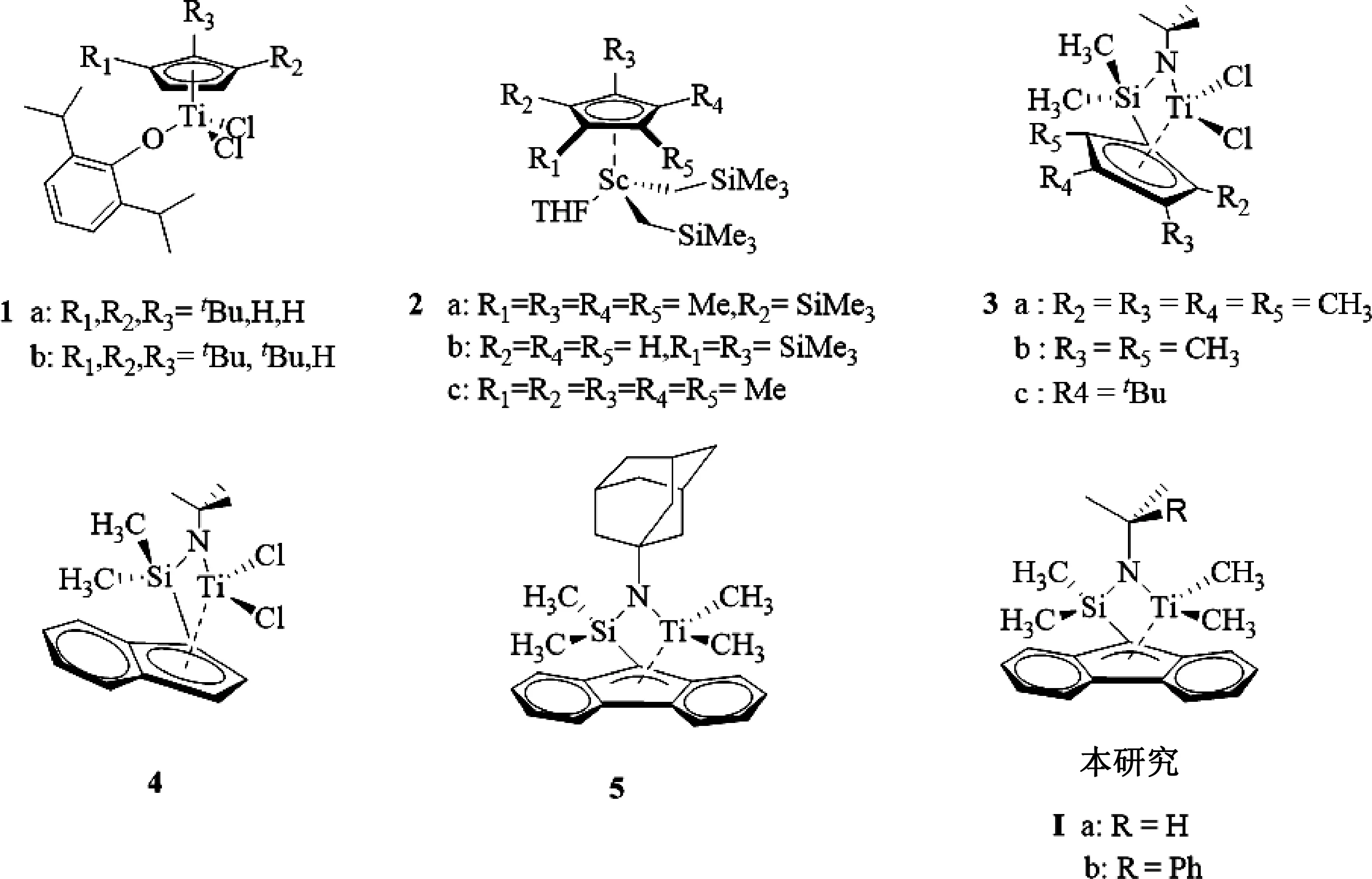
图1 单茂过渡金属催化剂
然而,上述聚合体系需使用钛配合物的200~2 000 当量比的MAO或MMAO对钛配合物进行活化,导致共聚产物中残留大量的铝化合物,影响其光学性能。综上,本文选用具有高活性的芴基胺基二甲基钛配合物 Ia和Ib,在1.2当量比的Ph3CB(C6F5)4活化下进行乙烯与NB及其NB衍生物双环戊二烯(DCPD)的共聚,研究了助催化剂对共聚性能的影响,如何通过改变聚合工艺条件对COC链结构加以调控进行了探索,并进一步表征探讨了COC的链结构与材料性能之间的关系,为镜头用COC材料的工业化生产提供理论基础。
1 试 验
由于整个实验过程对水和空气具有较强的敏感性,故所有操作都必须在高纯氮气的氛围下严格采用标准的Schlenk方法进行操作。在实验过程中所使用的高质量无水无氧有机溶剂都经过带有双净化柱的PS-MD-5溶剂纯化系统纯化后使用,参与反应的气体都是通过公司购买且纯度高达99.99 %并通过ZHD-20脱水柱与ZHD-20A脱氧柱进行纯化后参与反应。
1.1 原料
降冰片烯,99%,上海麦克林生化科技有限公司;双环戊二烯(99%),正丁基锂、甲基溴化镁、叔丁胺、二氯二甲基硅烷,均为分析纯,北京百灵威科技有限公司;甲苯、乙醚、正己烷、乙醇,均为分析纯,上海泰坦科技股份有限公司;高纯氮气(99.999%),乙烯(99.5%),上海申中特种气体有限公司。
1.2 仪器设备
NMR: Bruker AVANCE-600,德国 Bruker 公司制造,600 MHz,CDCl3为样品溶剂 (δ=7.26),TMS 为基准内标(δ=0)。
高温 GPC: PL-GPC 220,英国 Agilent Technolo-gies公司,150 ℃,溶剂为三氯苯(1,2,3-Trichloro-benzene)。
五路溶剂纯化系统: 美国 Innovative Technology公司。
DSC-Q2000: TA Instruments公司。
1.3 试验方法
配合物的制备:根据Cai等[9]文献合成了[t-BuNSiMe2Flu]TiMe2(Ia),合成路线如图2所示。
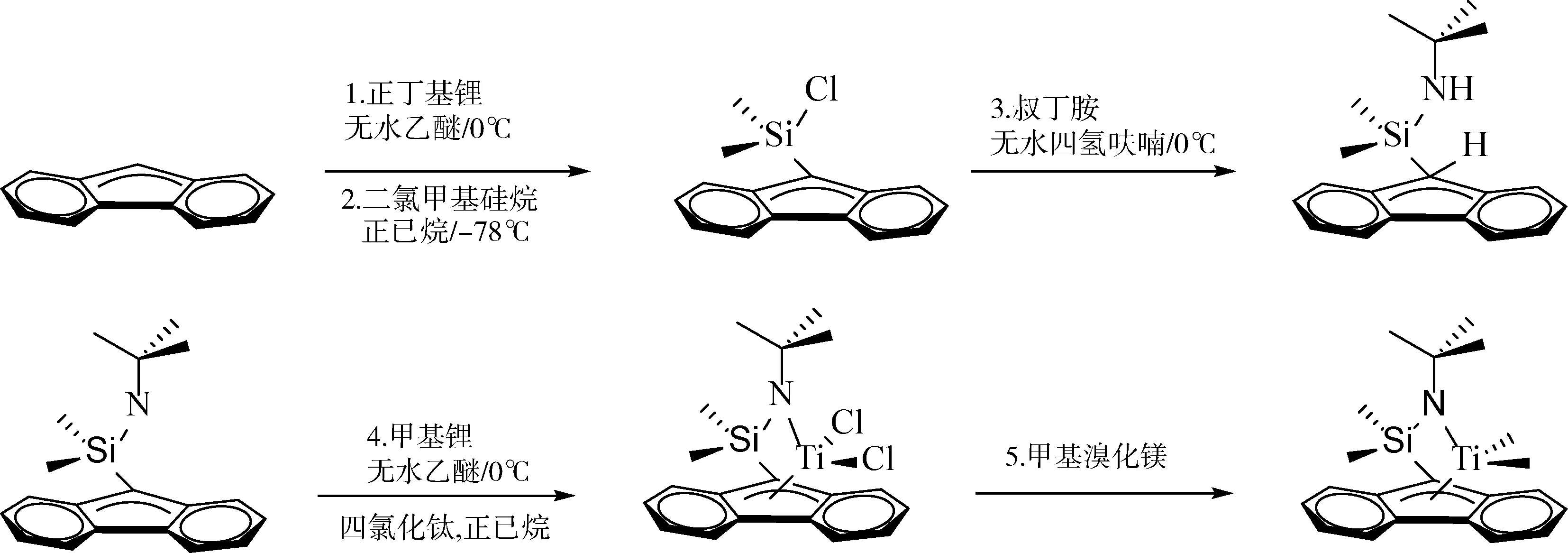
图2 [t-BuNSiMe2Flu]TiMe2(Ia)的合成路线
1) 取定量芴并用适量无水乙醚溶液溶解,并在0 ℃条件下加入正丁基锂溶液(1.1倍当量),常温条件下反应3 h。
2) 取4.5倍当量SiMe2Cl2用适量正己烷溶液溶解,在-78 ℃条件下将步骤1中橙红色溶液转移到烧瓶中,反应12 h。
3) 步骤2反应得到乳白色悬浊液,静置后将上层清液移出、抽干并称重。取与其同等当量的叔丁胺,并在0 ℃条件下逐滴加入正丁基锂(1.1倍当量)常温反应3 h。将之前上层清液溶解后转移至此烧瓶中反应6 h。用正己烷溶液充分清洗后离心抽干,得到橘黄色油状液体,即配体。
4) 取定量配体加入适量无水乙醚溶液溶解,在0 ℃条件下逐滴加入甲基锂(4.5倍当量)反应3 h,将上层清液与1.1倍当量四氯化钛正己烷溶液避光反应1 h。
5) 将步骤4溶液抽干并用正己烷溶液进行萃取,加入1.2倍当量的格式试剂反应1.5 h,过滤浓缩后放置于冰箱中重结晶,最终制得酒红色配合物晶体。
根据王华金等[10]文献合成了 [Flu(Me2)Si(Me2PhC)N]TiMe2(Ib)合成路线如图3所示。
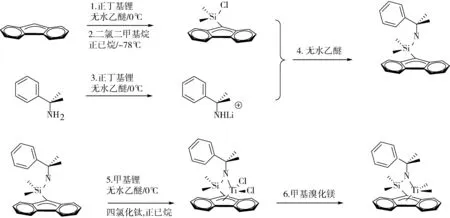
图3 [Flu(Me2)Si(Me2PhC)N]TiMe2(Ib)的合成路线
1) 取定量芴并用适量无水乙醚溶液溶解,并在0 ℃条件下加入正丁基锂溶液(1.2倍当量),常温条件下反应4 h。
2) 取4.5倍当量SiMe2Cl2用适量正己烷溶液溶解,在-78 ℃条件下将步骤1中橙红色溶液转移到烧瓶中,反应12 h。反应得到米黄色悬浊液,用正己烷溶液充分萃取后离心抽干,得到浅黄色固体。
3) 另取适量的异丙基苯胺用适量无水乙醚溶液溶解,并在0 ℃条件下加入正丁基锂溶液(1.1倍当量),恢复常温后继续反应3 h。
4) 将步骤2所得浅黄色固体用适量无水乙醚溶液溶解,并将其转移到步骤3中所得的胺盐溶液中,反应过夜。反应结束后,将溶液真空抽干并用正己烷萃取,得淡褐色配体。
5) 取定量配体加入适量无水乙醚溶液溶解,在0 ℃条件下逐滴加入甲基锂(4.5倍当量)反应3 h,将上层清液与1.1倍当量四氯化钛正己烷溶液避光反应1 h。
6) 将步骤5溶液抽干并用正己烷溶液进行萃取,加入1.2倍当量的格式试剂反应1.5 h,过滤浓缩后放置于冰箱中重结晶,最终制得深红色针状配合物晶体。
降冰片烯(或双环戊二烯)与乙烯共聚合:按照实验总体积取合适容积的单口烧瓶,对其进行抽真空回填高纯氮气三次以使体系内处于无水无氧环境,并用油浴锅控制体系处于稳定的实验温度条件下。
在高纯氮气气氛下依次往单口烧瓶中加入适量的甲苯、NB(或DCPD)单体,并将NB(或DCPD)/乙烯初始投料比严格控制在实验要求内。均匀搅拌15 min,待NB(或DCPD)单体在甲苯溶液分散均匀以及设定实验温度稳定后。
再次慢抽真空三次,迅速往聚合体系内回填乙烯气体,待气体流量计流速示数稳定后等待5 min使乙烯气体在甲苯中溶解至饱和状态。再将助催化剂与主催化剂用无水甲苯溶液充分溶解,依次加入至体系内,用秒表记录聚合反应时间。
待聚合结束后,立即使用适量的HCl/乙醇溶液终止,用无水乙醇充分搅拌洗涤聚合物两至三次,并抽滤使聚合物从溶液中完全分离后放置于真空干燥箱内恒温80 ℃干燥12 h。最终利用实验仪器对聚合物进行性能测定和分析。
1.4 分析测试
NMR测试:Bruker AsendTM 600 MHz核磁共振仪,称取约50 mg经纯化处理后的环烯烃共聚物产物放入核磁管中,并用氘代氯仿(CDCl3)作为溶剂。然后将样品密封在核磁试管中并在40 ℃条件下加热4~6 h,待其完全溶解后在室温条件下进行测试。
GPC测试:安捷伦公司的PL GPC-220高温凝胶色谱仪,称取约10 mg的环烯烃共聚物样品于样品瓶中并用1,2,4-三氯苯(TCB)溶剂在150 ℃条件下震荡溶解。待其完全溶解后使用滤枪移取部分溶液至测试瓶中,并将测试瓶放入自动进样器中待测,设定好测试参数后进行测试,测定结束后使用CIRRUS软件进行数据分析。测定时条件:温度:150 ℃;流动相:1,2,4-三氯苯(TCB);流速:1.0 mol/L;校准标样:窄分布聚苯乙烯。
DSC测试:TA Q2000差示扫描量热仪,称取5~10 mg环烯烃共聚物样品,将其封装在铝坩埚内并放置于仪器测试平台上,在惰性气氛环境以及预设的温度程序(升、降温速率:10 ℃/min;升温范围:40~250 ℃)下进行测定。
1.5 计算
催化剂催化聚合活性计算公式如下:
式中A指在单位时间内使用单位摩尔量催化剂所催化聚合得到COC的质量,kg/(mol·h);W聚合物为一定时间内催化聚合的聚合物质量,kg;M为实验中催化剂摩尔量,mol;t为聚合时间,h。
本实验中根据亨利定律计算出乙烯单体在甲苯溶液中的溶解度,以此计算出NB单体摩尔量,将NB/乙烯初始投料比保持不变来减小由于温度变化而产生的初始摩尔比差异,公式如下所示[11]:
式中C为乙烯在甲苯中的浓度,mol/L;P为乙烯压力,Pa;H0为亨利系数为0.001 75 mol/(L·Pa));ΔHL为乙烯在溶液中的溶解热(10 735 J/mol);R为气体常数,8.314 J/(mol·K);T为温度,K。
NB插入率BNB按以下计算公式[12]如下:
式中IC1/4、IC2/3、IC7值分别对应NB的C1/4、C2/3、C7特征峰的积分面积,ICH2值对应NB的C5/C6与乙烯CH2特征峰的积分面积之和。
DCPD插入率的计算公式[13]如下:
式中BDCPD为共聚产物中DCPD的插入率;IDCPD为对应谱图中峰值为3.00的特征峰积分面积;I乙烯为对应谱图中峰值从0.5~1.7的特征峰积分面积。
2 结果与讨论
2.1 钛配合物催化乙烯与降冰片烯共聚
通过采用芴基胺基二甲基钛类配合物Ia和Ib,以甲苯作为聚合反应溶剂,在固定共聚反应乙烯压力为101.325 kPa的条件下,探究了催化剂结构、助催化剂、温度条件以及乙烯/NB初始投料比等因素对NB与乙烯共聚反应的影响,结果如表1所示。
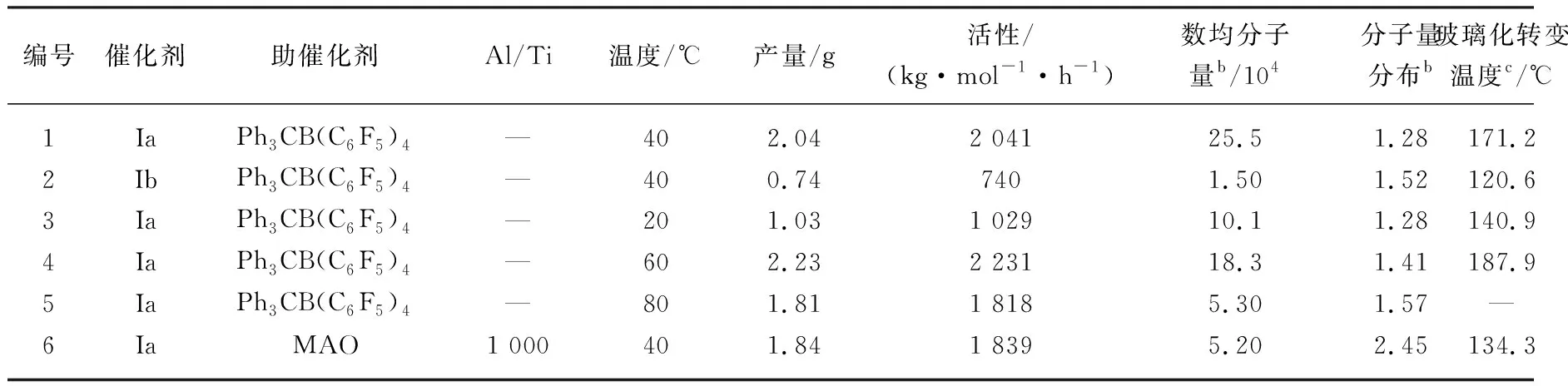
表1 钛配合物催化NB与乙烯共聚结果a
在相同的聚合条件下,Cs对称钛配合物的聚合活性Ia>Ib,其中含叔丁基胺的配合物Ia的共聚活性达到2 041 kg/(mol·h),原因可能是,在胺基上引入的苯环增加了金属中心空间位阻,导致NB单体的配位能力下降,从而使配合物的催化活性降低。在改变助催化剂条件下,当MAO作为助催化剂时即使Al/Ti比较高时,催化剂的聚合活性仍较低,只有1 839 kg/(mol·h),但再增加Al/Ti比时共聚活性则无明显增加,相比于Ph3CB(C6F5)4作为助催化剂时明显较低。在较高Al/Ti条件下,共聚产物的Tg仍然较低,而且聚合物分子量降低以及分子量分布变宽。可能是由于MAO中残留的三甲基铝不仅会与钛配合物络合形成双配位产物从而无法使钛配合物形成活性种使得其活性降低,还会增加链转移反应使得分子量降低(如图4所示)[14]。
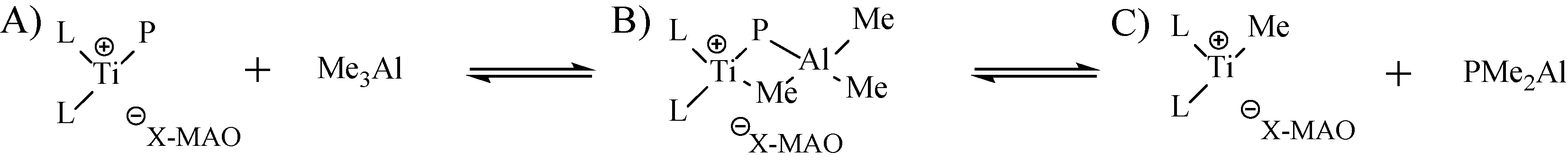
图4 MAO中残留的三甲基铝导致的副反应
在不同温度条件下,催化活性随着温度的升高催化剂活性先升高后降低。一般来说,茂金属催化剂在高于70 ℃的温度条件下容易发生催化剂茂环滑移以及配位状态改变等反应,而且随着温度的升高,乙烯在聚合溶液中的浓度下降,从而使催化活性降低。
从表1中可以看出,数均分子量(Mn)随着温度的升高先增加后减少,在40 ℃时分子量达到最大值,而后逐渐减少,到80 ℃时分子量降至最低。一方面,温度的升高缩短了诱导期延长了链增长反应时间,从而使得数均分子量增加。另一方面,温度升高使得乙烯单体在配位插入聚合链时更容易发生β-H 消除,促进了链转移反应,因此使得分子量急剧降低,分子量分布变宽。但即使如此,不同温度条件下的分子量分布仍稳定在1~1.6之间,这符合单活性中心催化剂的特点。
同时探究了不同乙烯/NB初始投料比条件对乙烯与NB共聚反应的影响,其结果如表2所示。

表2 钛配合物Ia在不同乙烯/NB初始投料比条件下催化NB与乙烯共聚结果a
在不同NB/乙烯初始进料比条件下,随着乙烯/NB初始投料比增加,共聚产物的Tg逐渐升高,如图5所示。可能是由于实验室内聚合反应采用的乙烯气体大气压为101.325 kPa,温度条件固定的情况下,乙烯单体溶解度不变。NB/乙烯初始投料比增加意味着NB单体浓度提升,因此其与活性中心碰撞的概率增大。从而配位插入到聚合产物中的NB单元增多,分子链刚性增加。分子链段移动更加困难,故在热流曲线中表现出Tg升高的现象。另外当NB/乙烯初始投料比过高时,初始聚合速率过快导致溶液浓度迅速上升,溶液难以搅动。乙烯气体受到传质阻力较大,溶解速率急剧下降,但体系内NB单体充足,故链结构中NB单元插入较快,从而使得COC产物的Tg升高。
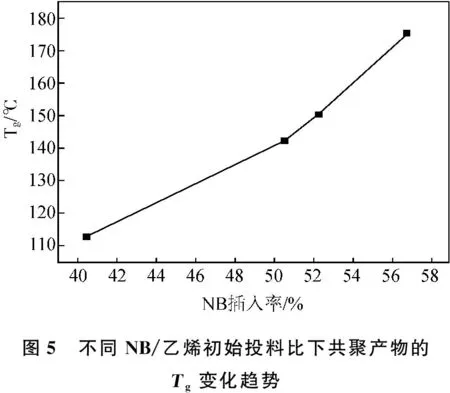
通过13C-NMR图谱中的NB碳的化学位移的变化可判断NB结构单元在共聚物中的二元组序列和三元组序列分布。结合表2和图5数据分析,Ia-Ph3CB(C6F5)4所得到的COC材料的热学性能即Tg与NB插入率之间呈现线性正相关关系,明显发现Tg随着NB插入率的增加而升高。另外从图中可以看出NB插入率无论是较高时还是较低时,碳谱中归属于二元组序列、三元组序列的峰强度都是较低的,说明聚合物主链中插入的NB单体多以孤立分布形式存在的。但随着链结构中NB单体插入率升高时,无论是二元组序列还是三元组序列的信号峰强度都是逐渐升高的,共聚产物的Tg迅速升高,故从2#~4#的Tg趋势呈现直线增长。可能是由于初始投料中NB单体较多,故连续插入的NB单体越多,同时使得催化活性和分子量增加。因此可以通过控制NB初始投料比来调控共聚产物中NB插入率,从而使制备得到的共聚产物的热学性能即Tg符合镜头用COC材料的性能要求。

图6 Ia-Ph3CB(C6F5)4体系催化共聚产物的13C-NMR谱图
2.2 钛配合物催化双环戊二烯与乙烯共聚
为进一步探究环烯烃共聚行为,利用钛配合物la-Ph3CB(C6F5)4催化体系催化DCPD与乙烯共聚,结果如表3所示。
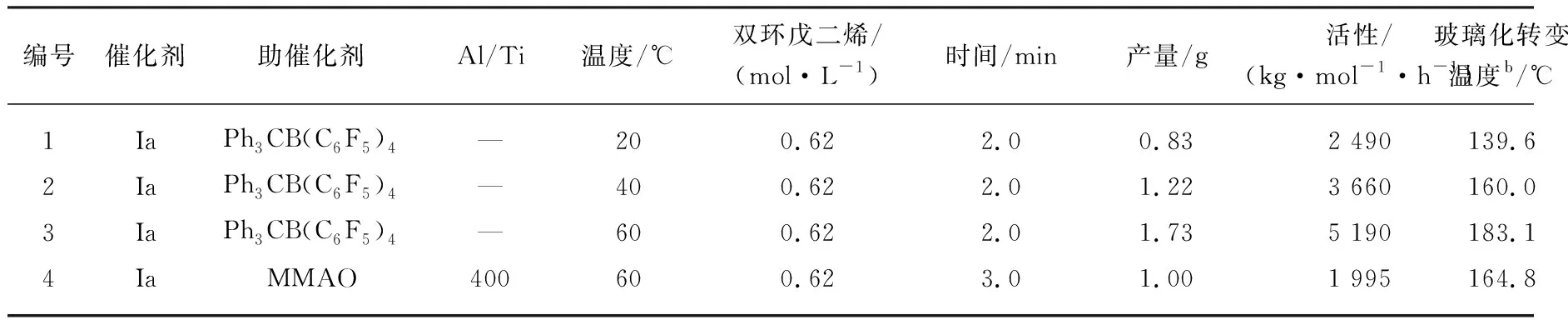
表3 钛配合物Ia催化DCPD与乙烯共聚结果a
通过表3发现,Ph3CB(C6F5)4作为助催化剂时聚合活性和共聚产物的Tg随着聚合温度的升高而升高。随着温度升高,催化剂对DCPD单体聚合能力增强,链中增多的DCPD使得链刚性增强,同时相对于表2中NB与乙烯共聚反应共聚产物的Tg也更高,这是由于DCPD除了双环结构外还存在着五元环和两个双键结构。较之于本课题组孙延杰等[12]利用MMAO作为助催化剂,该体系不仅在聚合活性上有较大提升,而且共聚产物的Tg也有明显提高。猜测其中原因可能是在MMAO中存在的未反应完全的三甲基铝引起副反应所造成的。
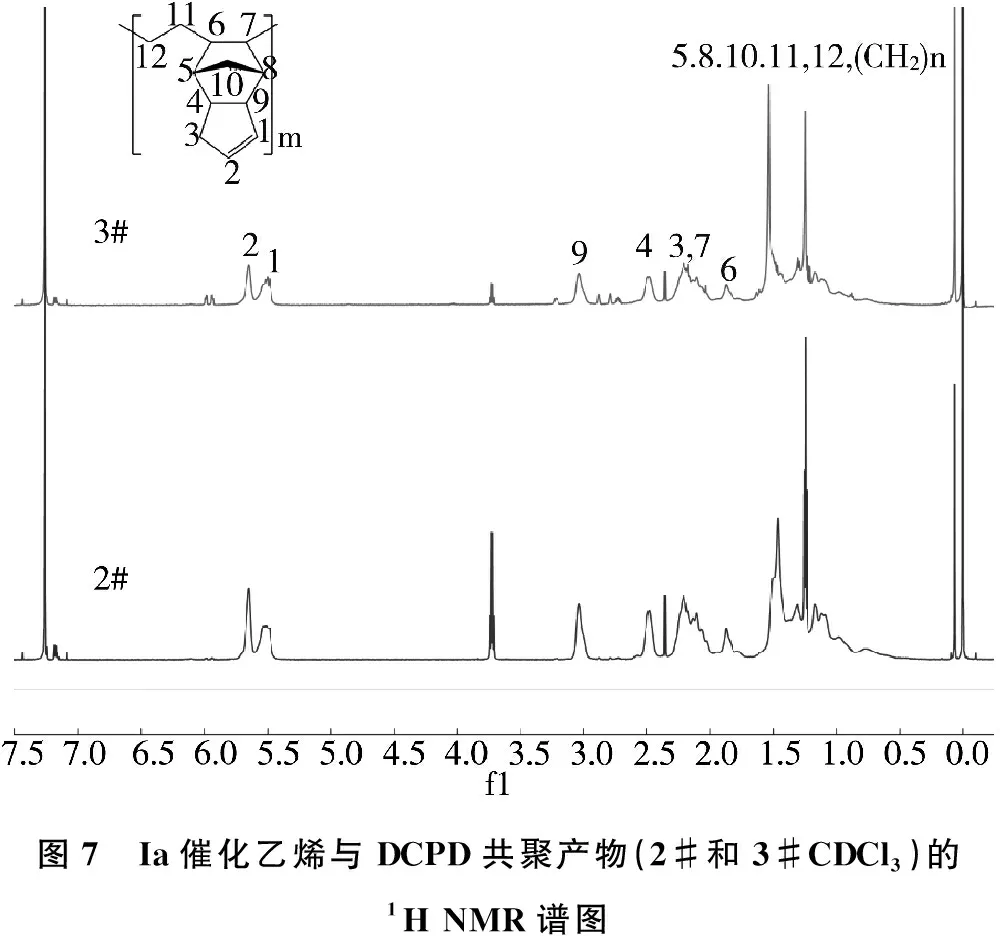
选取2#和3#所得共聚产物进行了NMR测试分析,并根据以下计算公式计算出2#和3#所得共聚产物中DCPD单体插入率分别为42.0%和46.7%。另外在孙延杰等研究中发现,所有共聚产物均发生了交联现象,可能是由于DCPD单体中另一个双键也参与了反应导致的,故本研究中减少了共聚反应时间,在谱图中可以看到出现了明显的DCPD中C1-C2 双键峰,这表明所得共聚产物中仍有部分双键保留在聚合物中且未反应。
3 结 语
本文以制备符合镜头用COC材料性能的光学高分子透明材料为导向,选用芴基胺基二甲基钛类配合物,研究了不同催化剂结构、助催化剂、温度以及共单体投料比等聚合条件对于乙烯与降冰片烯共聚反应的影响。
a)Cs-对称的[t-BuNSiMe2Flu]TiMe2(Ia)配合物在1.2当量比的Ph3CB(C6F5)4的活化下共聚活性最高达到2 040 kg/(mol·h),且对NB的共聚能力最强,NB的插入率在40.4%~56.7 %范围内可控。所得共聚产物的玻璃化转变温度(Tg)与NB的插入率呈线性关系,可在115~175 ℃范围内可控。
b) 同时探究乙烯与双环戊二烯(DCPD)的共聚结果发现,Ia-Ph3CB(C6F5)4体系对共聚表现出很高的活性,聚合温度为60 ℃时共聚合活性高达5 190 kg/(mol·h),共聚物中DCPD的插入率最高达46.7%。当两种共单体的插入率相同时,乙烯/DCPD共聚物的Tg高于乙烯/NB共聚物的Tg。
猜你喜欢
石油炼制与化工(2022年2期)2022-02-15 11:42:26
应用化工(2021年4期)2021-05-20 09:43:36
化工管理(2020年26期)2020-10-09 10:05:16
山东化工(2019年2期)2019-02-21 09:29:32
石油化工建设(2018年2期)2018-07-11 01:25:04
黄河之声(2016年24期)2016-02-03 09:01:52
火炸药学报(2014年3期)2014-03-20 13:17:39
断块油气田(2014年5期)2014-03-11 15:33:50
自动化博览(2014年9期)2014-02-28 22:33:35
无机化学学报(2014年9期)2014-02-28 17:33:05

- 合成技术及应用的其它文章
- ●一种服用阻燃聚酯纤维及其制备方法(申请号CN202011602594.5 公开日 2021-12-17 申请人 江苏恒力化纤股份有限公司)
- ●一种可降解混杂纤维透气毡、其制备方法及干燥方法(申请号CN202110512125.2 公开日 2021-08-17 申请人 宁波霓科新材料有限公司)
- ●一种带追踪剂再生聚酯纤维的制备方法 (申请号CN202111029704.8 公开日 2022-01-28 申请人 江苏恒力化纤股份有限公司)
- ●聚酯纤维及其制备方法(申请号CN202111237264.5 公开日 2022-02-08 申请人 苏州宝丽迪材料科技股份有限公司 )
- ●一种阳离子可染阻燃高强聚酯纤维的制备方法(申请号CN202111655078.3 公开日 2022-03-15 申请人 江苏新视界先进功能纤维创新中心有限公司)
- ● 一种改性聚酯纤维丝的制备方法(申请号CN202011595373.X 公开日 2021-11-02 申请人 盐城市恒固新材料科技有限公司 )