
初始钙-磷物质的量比值对超长羟基磷灰石纳米纤维微观结构的影响
2022-04-18 01:50王银川肖桂勇徐伟莉齐美丽颜文熙武延秋吕宇鹏
无机化学学报 2022年4期
王银川 肖桂勇*, 徐伟莉 齐美丽 颜文熙 武延秋 吕宇鹏*,
(1山东大学,材料液固结构演变与加工教育部重点实验室,济南 250061)
(2山东大学材料科学与工程学院,济南 250061)
(3山东交通学院交通土建工程学院,济南 250357)
(4生物医用材料改性技术国家地方联合工程实验室,德州 251100)
0 引言
羟基磷灰石(HA,Ca10(PO4)6(OH)2)作为脊椎动物骨骼和牙齿中的主要无机成分,因其良好的生物活性、生物相容性、骨诱导性和骨传导性,在载药、蛋白分离、骨缺损和组织工程修复等领域具有广泛应用[1⁃2]。但传统HA陶瓷因脆性大、韧性差的缺陷使其局限应用于非动态载荷领域,极大地限制了HA的临床应用价值[3]。研究表明纤维复合陶瓷中的纤维可通过吸收断裂能并防止裂纹快速扩展来有效增强陶瓷的力学性能[4]。然而,目前常用的C、Al2O3、ZrO2、SiC等生物惰性纤维虽然有效提高了复合材料的断裂韧性,但会导致复合陶瓷的生物相容性降低,甚至可能导致细胞毒性[5⁃6]。因此,制备高长径比的HA纤维实现HA陶瓷的自增韧,成为目前的研究热点[7⁃8]。
尽管已有水热法[9]、均相沉淀法[10]、水热均相沉淀法[11⁃12]和溶剂热法[13]等[8]多种方法用于合成HA纤维,但制备超高长径比和柔韧的HA纤维目前仍是一个巨大的挑战。晶体生长与起始物密切相关,其中表面活性剂可有效调控晶体形貌和尺寸[3,14]。此外,晶体的形核率和生长还取决于溶液的过饱和度,而过饱和度与初始钙−磷物质的量比值(nCa,0/nP,0)直接相关。初始Ca2+浓度恒定时,过低的nCa,0/nP,0会导致溶液的过饱和度过高,生成大量HA小晶核,并促使其沿a轴生长,进而影响HA纤维的微观结构[3]。因此,选择合适的、可调控HA晶体沿c轴择优生长的表面活性剂和适宜的nCa,0/nP,0,有望合成超长柔韧的HA纤维。研究表明具有合适链长脂肪酸分子的羧基在共面时,羧基间的间距与HA晶体(100)和(001)晶面上共面的Ca2+间距相匹配,从而诱导HA晶体的择优生长取向[15⁃16]。并且脂肪酸作为有机弱酸,在一定pH范围内可作为pH响应性分子维持溶液的pH值稳定[17⁃18]。
我们以油酸作为表面活性剂,制备出具有高柔韧性和高长径比的HA纳米纤维,探讨了溶剂热条件下不同nCa,0/nP,0对HA纳米纤维微观结构的影响。并基于不同nCa,0/nP,0下HA形貌和结构演变推测出相应的影响机理,以期为不同脂肪酸作为表面活性剂制备超长HA纳米纤维时参数的选择提供依据。
1 实验部分
1.1 实验材料
实验所用无水氯化钙(CaCl2,AR)、氢氧化钠(NaOH,AR)、二水合磷酸二氢钠(NaH2PO4·2H2O,AR)、无水乙醇(C2H5OH,AR)统一购自国药集团化学试剂有限公司,油酸(C18H34O2,AR)购自上海阿拉丁生化科技股份有限公司,无需进一步提纯即可使用。
1.2 一维羟基磷灰石纳米纤维产物的制备
采用溶剂热法制备得到一维纳米结构的HA产物。首先,在室温下将12.00 g油酸、11.00 g无水乙醇和10 mL去离子水混合得到溶液A。随后依次向混合液 A 中逐滴加入 CaCl2水溶液(0.2 mol·L−1,20 mL)和NaOH水溶液(1.25 mol·L−1,20 mL)得到反应液B。最后向反应液B中逐滴加入20 mL含一定量NaH2PO4·2H2O的水溶液得到混合物C。本研究中nCa,0/nP,0值分别设置为0.6、0.8、1.0、1.2、1.4、1.6、1.8、2.0。上述过程均在密闭体系且剧烈的磁力搅拌下进行。之后将混合物C转移到100 mL水热反应釜中,控制填充度为80%,在180℃下恒温24 h。待反应釜冷却至室温后,将制备得到的产物用无水乙醇和去离子水清洗3次,最终离心、干燥。
1.3 表 征
利用RIGAKU UltimaⅣ型X射线衍射仪(XRD)检测产物的相组成,辐射源为λ=0.154 18 nm的铜靶(CuKα),加速电压和加速电流分别为40 kV和40 mA,扫描范围为10°~60°。利用配有牛津X⁃max 80型X射线能谱仪(EDS)的JSM⁃7800F型场发射扫描电子显微镜(FESEM)分析检测产物表面形貌和微观区域的化学成分,因样品不导电,测试前将样品分散在无水乙醇中,然后滴在铝箔上,待干燥后进行喷铂处理。利用Nexus 670型傅里叶变换红外光谱仪(FTIR)分析产物官能团组成,扫描范围为4 000~400 cm−1,测试前将约0.2 mg样品与约20 mg溴化钾研磨混合后压片。
所制备产物的元素含量基于EDS结果获得,晶粒尺寸(Dhkl)根据德拜−谢乐公式计算得到[19⁃20],结晶度(XC)可由以下公式确定[21]:
XC=1−V112/300/I300
式中,V112/300为(112)和(300)晶面衍射峰之间峰谷的衍射强度;I300为(300)晶面衍射峰的衍射强度。
2 结果与讨论
2.1 nCa,0/nP,0对HA物相与化学成分的影响
图1是不同nCa,0/nP,0值下制备产物的XRD图及相应的晶粒尺寸、c/a值。如图1a所示,不同nCa,0/nP,0对产物的物相无明显影响,所制备的产物为HA(PDF No.09⁃0432)。nCa,0/nP,0=2.0~1.4时,所制备的产物在32.5°附近的漫散包表明产物中存在非晶磷酸钙(ACP);随着nCa,0/nP,0值的降低,4种产物的(002)晶面衍射峰的相对强度逐渐降低,且nCa,0/nP,0=1.4的产物的漫散包最强值从32.3°变为33.1°,表明随着nCa,0/nP,0值的降低,HA晶体的择优生长取向由a轴向c轴转变且ACP含量逐渐降低。nCa,0/nP,0=1.2时,衍射峰由漫散包突变为较尖锐的衍射峰,说明该nCa,0/nP,0值为产物主要组成由非晶态向晶态转变的转折点且产物的形貌可能发生明显的改变。随着nCa,0/nP,0值的进一步降低(nCa,0/nP,0=1.0~0.6),各产物的最强峰均为(300)晶面,说明低nCa,0/nP,0值是利于HA晶体沿着c轴择优生长;但结合表1和图1b可以看出,nCa,0/nP,0=1.0时所得产物具有最高的结晶度和晶粒尺寸,虽然nCa,0/nP,0=0.8时产物具有最大的c/a值,但是其晶粒尺寸已急剧减小。以上结果表明,nCa,0/nP,0=1.0为溶剂热法制备一维HA纳米纤维的最佳值,较低的nCa,0/nP,0有利于HA晶体沿着c轴择优生长,但过低的nCa,0/nP,0值会导致生成的HA晶体具有较小的晶粒尺寸且同时沿c轴和a轴生长。

表1 不同nCa,0/nP,0值下制备产物的合成参数和表征结果比较Table 1 Comparison of synthesis parameters and characteristics of as⁃prepared products synthesized with various nCa,0/nP,0values

图1 不同nCa,0/nP,0值下制备产物的(a)XRD图、(b)晶粒尺寸和c/a值Fig.1 (a)XRD patterns,(b)crystallite sizes,and c/a values of as⁃prepared products synthesized with various nCa,0/nP,0values
通过FTIR进一步研究nCa,0/nP,0对HA纳米纤维化学成分的影响(图2)。如图2所示,3 422 cm−1处的吸收峰是吸附在样品表面的游离水峰[22];3 566和631 cm−1处对应 HA 中 OH−的伸缩振动吸收峰[23];1 096、1 071、1 027、605 和 560 cm−1处对应 HA 中PO43−的振动吸收峰[24]。另外,3 007 cm−1处对应油酸根离子中=CH的特征峰,2 923和2 853 cm−1处对应油酸根离子中—CH2—的振动峰,1 557和1 465 cm−1处对应油酸根离子中—COO−的特征峰[25]。以上吸收峰的存在表明样品表面吸附有油酸根离子。此外,各产物中属于油酸根的特征峰峰强随nCa,0/nP,0值的降低亦逐渐降低,可能是nCa,0/nP,0的降低会导致各样品表面油酸根离子附着量逐渐减少。
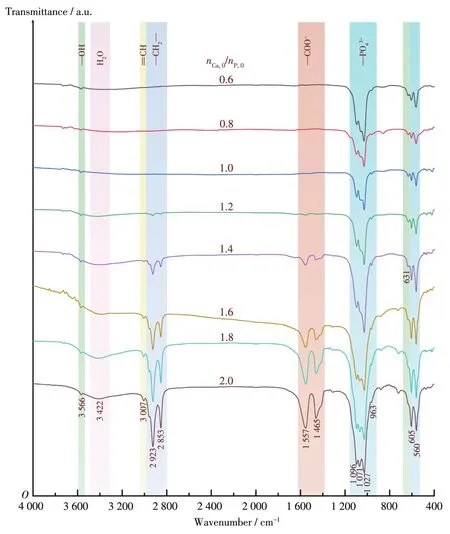
图2 不同nCa,0/nP,0值下制备产物的FTIR谱图Fig.2 FTIR spectra of as⁃prepared products synthesized with various nCa,0/nP,0values
2.2 nCa,0/nP,0对HA形貌的影响
图3和表2是不同nCa,0/nP,0值下制备产物的FESEM图及相应的EDS元素分析结果。如图3所示,随着nCa,0/nP,0值的降低,产物的形貌逐渐由绳结状向绳结状与纤维状共存,再到超长柔韧纳米纤维,最后到纳米纤维与纳米针束状纤维共存演变。nCa,0/nP,0=2.0时,从嵌入的高倍图中可以看出,绳结状产物表面附着有少量纳米颗粒(图3a),结合表2元素含量和相应的XRD结果(图1a)推测,绳结状产物可能为ACP、沿a轴择优生长的HA和油酸根离子组成的混合物。nCa,0/nP,0=1.4~1.8时,纤维形貌随nCa,0/nP,0值降低逐渐增多,而绳结状形貌逐渐减少,但仍可观察到两者表面均附着有纳米颗粒(图3b~3d)。nCa,0/nP,0=0.8~1.2时,绳结状形貌彻底消失,产物由高长径比(>10 000)的HA纳米纤维组成(图3e~3g),纤维呈任意角度弯曲而不断裂,表明纤维具有较高的柔韧性[17]。但nCa,0/nP,0降至0.6时,产物主要形貌变为纳米针束状纤维,并存在少量HA纳米纤维。此外,结合相应的FTIR结果可知,产物完全由纤维组成时油酸根的特征峰峰强几乎消失,这可能是因为组成绳结状产物的ACP和HA纳米颗粒具有较纤维状HA更大的比表面积,从而能附着有较多的油酸根离子。而产物中ACP和HA纳米颗粒的量随nCa,0/nP,0值降低而减少,导致各产物中属于油酸根的红外特征峰峰强随nCa,0/nP,0降低而逐渐降低。

图3 不同nCa,0/nP,0值下制备产物的FESEM图Fig.3 FESEM images of as⁃prepared products synthesized with various nCa,0/nP,0values

表2 不同nCa,0/nP,0值下制备产物的EDS元素分析结果Table 2 Compositions of as⁃prepared products synthesized with various nCa,0/nP,0values derived from EDS analysis
表1中产物的nCa/nP结果表明,HA产物的nCa/nP值随nCa,0/nP,0值的降低而降低。具有柔韧纤维状形貌的HA产物的nCa/nP=1.11~1.35,远低于HA的理论nCa/nP值(1.67)。这可能归因于油酸诱导HA晶核沿c轴择优生长,导致HA晶体中出现钙缺陷[26],使其nCa/nP降低。然而当产物的nCa/nP≈1.67时形貌为非晶绳结状。以上结果表明,溶剂热条件下油酸诱导合成具有超长柔韧性缺钙型HA纳米纤维的nCa,0/nP,0的最佳范围为0.8~1.2,且可通过调控nCa,0/nP,0实现对HA产物结晶度和晶粒尺寸的控制。
2.3 nCa,0/nP,0对HA纳米纤维微观结构影响机理
晶体的形核和生长速率与溶液的过饱和度密切相关。低过饱和度的溶液中形核率低,有利于更大晶粒尺寸的晶体形成[3]。根据以下公式[27],HA的过饱和度(S)与溶液中游离的Ca2+、PO43−和 OH−离子浓度直接相关;当OH−离子浓度恒定时,nCa,0/nP,0可直接影响反应溶液的过饱和度,进而调控HA晶体的形核与生长。

式中,α为离子的浓度;Ksp为HA的溶度积(特定温度下为一常数)。此外,油酸作为表面活性剂不仅可以有效诱导HA晶体沿着c轴择优生长,也可以作为pH响应性分子调控反应溶液的pH值,使其维持稳定,并且油酸参与反应后生成的油酸钙可作为钙源在反应过程中缓慢释放Ca2+离子。因此,在油酸分子的作用下,nCa,0/nP,0对HA晶体微观结构的影响需要系统的探究,以期能合成具有更高长径比和柔韧性的HA纳米纤维。基于上述表征分析和相关报道,不同nCa,0/nP,0对HA纳米纤维的形核及生长的影响可分为以下3个阶段(图4):
2.3.1 阶段Ⅰ:反应初始
如图4中stageⅠ所示,油酸分子与NaOH反应生成油酸钠,随后与游离的Ca2+离子结合生成油酸钙。油酸钙在反应中作为前驱体和钙源[28]。HPO−24离子与OH−离子反应生成HPO2−离子[17],少量 HPO2−44离子继续与OH−离子反应生成PO43−离子。
2.3.2 阶段Ⅱ:HA形核和晶体生长
如图4中stageⅡ所示,随着溶剂热反应的进行,油酸钙开始缓慢且持续地释放Ca2+离子,同时油酸根离子水解释放OH−离子。大量HPO−、HPO2−244离子与OH−反应分别生成HPO42−和 PO43−离子。当反应溶液中的游离Ca2+、PO43−和OH−离子达到HA形核的过饱和度后,少量HA晶核在溶液中直接形核,大部分游离Ca2+和PO43−离子在离子间库仑力的作用下生成ACP纳米粒子[29]。随着反应的继续进行,直接形核的HA晶核开始长大,另外HA也开始在ACP表面异质形核。此外,相关研究表明HA从反应溶液中形核析出时表面具有负的ζ电位[30]。因此,在生长初期,可能由于HA晶核表面的负电势抑制了油酸根离子与HA表面的钙位点结合。HA晶核在生长初期无油酸根离子诱导的作用下,为了维持体系能量最低,呈现出沿着a轴择优生长的趋势。伴随着HA晶核的不断长大,其表面自由能不断升高,在HA晶体暴露出带正电的面积较大的晶面时,反应液中的油酸根离子吸附在该晶面的钙位点上,限制了PO43−离子与平行于c轴方向的晶面上的钙位点结合,促使HA晶体只能沿着c轴择优生长。此外,研究表明HA晶体沿c轴生长较其沿a轴生长所需的能量更多[31],高温、高压的溶剂热环境为HA晶体沿c轴生长提供了所需的能量条件,最终生长成为短的单根HA纤维。

图4 不同nCa,0/nP,0值下制备的产物形成过程示意图Fig.4 Schematic illustration of the formation process of as⁃prepared products synthesized with various nCa,0/nP,0values
当nCa,0/nP,0=2.0时,反应溶液中的PO43−离子浓度极低,虽溶液的过饱和度低,但磷源的不足导致只有少量HA晶核能生长成沿a轴择优生长的小晶粒,大量HA晶核则多数停止晶体生长。1.4≤nCa,0/nP,0<2.0时,反应溶液中的过饱和度仍然较低,溶液中的PO43−离子已能满足部分HA晶核在油酸的诱导下沿着c轴择优生长为较短的HA纳米纤维,而部分HA仍是以低结晶小晶粒的形式存在或仍残留部分ACP。0.8≤nCa,0/nP,0<1.4时,反应溶液中的过饱和度处于适当的范围,在此过饱和度下生成的HA晶核不仅较少,溶液中的PO43−离子也能满足HA晶核生长为具有较大晶粒尺寸的HA纳米纤维的需求。0.6≤nCa,0/nP,0<0.8时,溶液过饱和度较高,导致了大量HA晶核的生成,最终生长成为具有较小晶粒尺寸的针束状HA纳米纤维;此外,因溶液中过高的PO43−离子浓度,PO43−离子可以充分地与HA表面上的钙位点结合,抑制了油酸根离子在HA表面的吸附,导致nCa,0/nP,0=0.6时制备产物的D211>D300。
2.3.3 阶段Ⅲ:反应结束
在溶剂热过程中,油酸根离子会吸附在产物的表面,而油酸根离子中的烷基链与极性溶剂互不相容[28]。因此,当溶剂热反应结束,将产物转移至极性溶剂(水、乙醇)中时会自组装成为高度有序的形貌。如图4中stageⅢ所示,HA和ACP纳米颗粒在“油酸根离子−极性溶剂”的作用下自组装成为绳结状形貌;短的HA纳米纤维自组装成为超长高柔韧性HA纳米纤维;HA纳米针自组装为HA纳米针束状形貌。
3 结论
nCa,0/nP,0对HA纳米纤维的形貌和结构特征具有显著影响。随着nCa,0/nP,0值降低,产物由非晶绳结状(nCa,0/nP,0=2.0)向高结晶超长柔韧的HA纳米纤维(0.8≤nCa,0/nP,0<1.4)转变,最终为具有较低结晶度、小晶粒尺寸的针束状HA纳米纤维(0.6≤nCa,0/nP,0<0.8)。HA晶体的择优生长取向亦随nCa,0/nP,0的降低由a轴转变为c轴,但过高和过低的nCa,0/nP,0均会抑制油酸的诱导作用,并且过低的nCa,0/nP,0会导致HA晶体同时沿a轴和c轴生长。nCa,0/nP,0=0.8和1.2分别为产物微观结构发生转变的关键点;合成HA纳米纤维的最佳nCa,0/nP,0=0.8~1.2,其中nCa,0/nP,0=1.0时的HA纳米纤维具有最大晶粒尺寸和结晶度。以上结果可能为溶剂热法合成HA纳米纤维提供一些新的思路。
猜你喜欢
生物技术进展(2022年5期)2022-10-11
煤炭与化工(2022年5期)2022-06-17
中国种业(2021年1期)2021-12-05
人工晶体学报(2021年10期)2021-11-26
粉末冶金技术(2021年3期)2021-07-28
表面工程与再制造(2019年3期)2019-09-18
当代陕西(2018年9期)2018-08-29
环球时报(2018-03-01)2018-03-01
科技资讯(2017年24期)2017-09-15
