
计算机模拟亚甲基蓝与牙龈卟啉单胞菌部分蛋白的分子对接
2022-04-14 10:13袁临天马利沙刘润园张栌丹王贵燕王宇光
北京大学学报(医学版) 2022年1期
袁临天,马利沙,刘润园,齐 伟,张栌丹,4,王贵燕,5,王宇光△
(1.北京大学口腔医学院·口腔医院综合科,国家口腔疾病临床医学研究中心,口腔数字化医疗技术和材料国家工程实验室,口腔数字医学北京市重点实验室,北京 100081; 2.北京大学口腔医学院·口腔医院口腔医学数字化研究中心,北京 100081; 3. 大连医科大学口腔医学院牙体牙髓科,辽宁大连 116044; 4.北京大学口腔医学院·口腔医院门诊部,北京 100081; 5. 北京大学口腔医学院·口腔医院儿童口腔科,北京 100081)
牙周病是由细菌毒力和宿主防御能力失衡引起的口腔常见疾病,可引起牙列缺损、牙列缺失[1]、种植体周围感染和脱落[2-3],甚至影响全身健康[4-5]。牙龈卟啉单胞菌(Porphyromonasgingivalis,Pg)是牙周疾病中的关键致病菌之一[6],可通过FimA、Mfa等蛋白的作用黏附于宿主,增加其侵袭性和抵抗性,不易被免疫系统和药物清除[7-8]。
光动力抗菌疗法(antimicrobial photodynamic therapy,aPDT)是一种将外源性光敏剂与细菌孵育后,在有氧的条件下通过光激发与细菌结合的光敏剂,进而产生活性氧,最终杀灭微生物的方法[9]。光敏剂在光动力治疗中起决定性的作用[10],目前研究较多的光敏剂根据其结构分为:四吡咯结构光敏剂、天然光敏剂和合成染料光敏剂。亚甲基蓝是阳离子合成染料光敏剂,带正电荷,对革兰氏阳性细菌和革兰氏阴性细菌均有亲和力,但一般认为光敏剂对革兰氏阴性细菌的亲和力弱于革兰氏阳性细菌[11-13]。然而,光敏剂光照后产生活性氧簇所作用的范围有限,仅能对邻近生物大分子产生影响[14-16],因此,光敏剂是否能与Pg的蛋白结合,是亚甲基蓝能否杀灭Pg的先决条件。有研究表明亚甲基蓝等光敏剂在光照后能降低FimA基因在Pg中的表达[17],但光动力杀菌在蛋白层面的相关研究较少。蛋白质是生命活动功能的执行者,如何高效研究光敏剂对蛋白质的作用,值得进一步探索。
近年来,生物信息学和计算机模拟分子模型被引入到药物的研发中,如Pourhajibagher等[18]通过多个在线数据库(I-TASSER、Interpro、STRING、Protter等)进行FimA蛋白的拓扑结构和骨架预测、蛋白质间相互作用网络分析,认为FimA蛋白具有7个配体结合位残基和1个可能与阳离子光敏剂甲苯胺蓝结合的活性位点残基。此外,有研究利用ProSA数据库研究光敏剂吲哚菁绿与Pg精氨酸牙龈蛋白酶相互作用,发现该蛋白位于Pg的外部,具有9个域和17个配体,可能是与阴离子光敏剂吲哚菁绿的相互作用有关,但这些研究均没有进一步探讨计算相关蛋白的活性位点与光敏剂的结合能和可能的三维结合方式[19]。
当前,有多种通过计算模拟药物与蛋白靶点对接的方法,其中AutoDock是经典的开源软件,用于小分子药物配体与大分子蛋白受体的对接和虚拟筛选[20],探究药物与蛋白或聚合物的作用[21-23]。2017和2020年分别有研究将其引入光动力和声动力杀灭肿瘤的实验,探究了光敏剂维替泊芬与人转录共激活因子相关蛋白、卟啉钠与人血清白蛋白的亲和程度及三维结合位点[24-25],但该方法在光动力杀菌的研究中少有报道。
本文结合上述的两种方法,通过靶点网络预测数据库与AutoDock分子对接软件研究光敏剂亚甲基蓝与Pg蛋白的作用靶点、结合能,模拟光敏剂与目的蛋白的三维空间结合,提高筛选光敏剂与细菌蛋白作用靶点的效率,为光动力作用机制的探究提供新的方法。
1 材料与方法
1.1 靶点网络预测
1.1.1收集Pg致病相关蛋白 在Uniprot数据库(https://www.uniprot.org)和RCSB PDB数据库(https://www.rcsb.org/)中获取Pg的蛋白名称(Pg菌株为W83)。
1.1.2获取亚甲基蓝有效成分结构 为获得光敏剂亚甲基蓝的准确成分,从以下4个数据库SciFinder(http://scifinder.cas.org)、PubChem(https://pubchem.ncbi.nlm.nih.gov/)、ChemSpider(http://www.chemspider.com)、Chemical Book(http://www.chemicalbook.com/)中筛选并对比亚甲基蓝的结构,并用ChemBioDraw软件绘制,将绘制完成的亚甲基蓝文件保存为*.mol2格式和*.sdf格式。
1.1.3预测亚甲基蓝潜在作用靶点 为寻找光敏剂亚甲基蓝是否有潜在的作用靶点,研究将第1.1.2小节所获得的*.mol2文件导入PharmMapper数据库(http://www.lilab-ecust.cn/pharmmapper/)进行靶点预测。将获得的亚甲基蓝靶点信息与UniProt上的蛋白质信息比对,不限定物种,将检索得到的所有蛋白校正为其官方名称。经过上述操作,获得有效成分预测靶点的信息,这些靶点来自于所有物种,采用可视化软件Cytoscape(版本: 3.7.0)构建网络。
1.1.4筛选蛋白质相互作用数据 String数据库(https://string-db.org/)是已知和预测蛋白质-蛋白质相互作用的数据库。为寻找光敏剂亚甲基蓝潜在的作用靶点与Pg的关系,从String 数据库中搜索蛋白质-蛋白质相互作用(protein-protein interactions, PPI)数据,其物种仅限于“Pg”,其中设置置信度为大于0.4,绘制PPI数据图,网络中的节点表示活性成分、靶点或作用通路等。
1.2 分子对接验证
使用AutoDock软件 (版本4.2,来源:http://autodock.scripps.edu/downloads)进行分子对接,该软件基于C语言开发,采用拉马克遗传算法来寻找配体与受体最佳的结合状态。本文中配体是光敏剂亚甲基蓝,受体是Pg的多种蛋白。AutoDock软件中的第一个程序AutoGrid用于相关能量的计算,第二个程序AutoDock用于分子对接,AutoDock 对接的基本流程如下。
1.2.1处理受体蛋白 本研究将亚甲基蓝与多种Pg已解析结构的蛋白进行对接。以FimA蛋白为例,从RCSB PDB数据库中搜索Pg的FimA蛋白编号4Q98,下载4Q98.pdb文件,或搜索“FimA w83“亦可找到4Q98.pdb文件,受体蛋白处理流程为:加氢→去水→更改原子类型。蛋白的分子结构在解析时是无法扫描氢原子的,因此“加氢”是分子对接的第一步骤。水被认为是分子对接时不需要的杂物,因此需要“去水”的步骤来避免分子对接时水对结果的影响。更改原子类型处理完后,将处理后的受体蛋白文件另存成*.pdbqt格式。
1.2.2处理配体小分子 处理配体小分子之前,首先关闭受体蛋白文件。从“第1.1.2小节有效成分结构获取”部分得到的亚甲基蓝分子结构载入配体文件,配体需经过处理,以包含可旋转结构进行半柔性对接。配体小分子处理流程如下:(1)导入配体,将第1.1.2小节中通过数据库下载且用软件绘制完成的亚甲基蓝*.sdf文件导入到软件;(2)观察结构,区分配体的刚性部分和非刚性部分;(3)配体定点,确定并设定中心点,作为调整视野角度时的参考;(4)固定配体,将配体可旋转结构转为不可旋转结构;(5)输出文件,将处理后的亚甲基蓝配体文件另存为*.pdbqt格式。
1.2.3设置受体和配体的网格框 为确定和限定软件对受体和配体的三维分子对接的范围,载入蛋白质受体文件与亚甲基蓝配体文件,设置受体网格框。需要注意的是,网格框不要触碰到配体的界面。设置受体和配体网格框流程如下:(1)重启软件,并载入由第1.2.1小节所得的已处理蛋白质*.pdbqt格式文件和由第1.2.2小节所得的亚甲基蓝配体小分子*.sdf格式文件;(2)设置参数,调整网格框(软件中gridbox)的大小和位置;(3)输出文件,将以上已设置受体和配体网格框的文件另存为*.gpf格式。
1.2.4用Autogrid程序计算结合位点能量 AutoDock软件中的 autogrid 子程序主要负责格点中相关能量的计算,基本操作如下:(1)输入指令,选中autogrid,按住Shift键,右击“在此处打开命令窗口”,在命令窗口内输入运行指令“autogrid4.exe-pdock.gpf”,点击回车键即可见运行状态;(2)矫正参数,根据运行状态的提示内容校正参数;(3)输出文件,将进行能量计算完成后的文件输出并另存为dock.gpf格式。操作说明:这里需要注意的是,所有的文件及文件夹命名均不能有中文和空格。
1.2.5分子对接 在AutoDock软件中的autodock子程序负责构象搜索及分子对接。基本操作如下:(1)载入文件,载入第1.2.1小节和第1.2.2小节所得的*.pdbqt格式的受体蛋白文件和亚甲基蓝配体文件,还有第1.2.3小节的*.gpf格式的网格框的文件;(2)分子对接,对接程序运行完成后输出*.dpf格式文件,打开*.dpf格式文件,查看分子对接的参数;(3)分子对接完成后另存为*.dlg格式文件;(4)导入由第1.2.2小节所得的*.pdbqt格式的亚甲基蓝配体文件和本步骤所得的*.dlg格式的小分子文件,分析对接结果。操作说明:在完成能量计算后,载入由第1.2.2小节所得已处理的*.pdbqt格式的亚甲基蓝配体文件,之后需要确定两个三维结构中哪一个是配体,哪一个是受体,并需要设置分子对接的配体。此外,使用默认参数,设置分子对接参数,进行分子对接。在完成分子对接后,输出*.dpf格式文件,查看相关的对接参数。当命令运行/对接完成后,会在默认文件夹形成*.dlg格式文件。此时再导入dock.dlg文件和受体蛋白的*.pdbqt文件,导入后设置相关参数,将小分子加载到蛋白中。自此,分子对接所有步骤均已完成。
2 结果
2.1 成分-靶点网络分析
本研究将三维结构模式图提交到可以进行药效团结构靶点分析的PharmMapper数据库,进行潜在靶点的反向筛选,最终得到亚甲基蓝潜在靶点,共268个。
图1为Cytoscape软件构建的药物与靶点可视化网络(仅显示268个靶点中的40个靶点),其中红色代表亚甲基蓝,蓝色代表亚甲基蓝的靶点。直接进行亚甲基蓝的反向靶点筛选发现亚甲基蓝与细菌结合的靶点较多,需要通过蛋白质相互作用方法研究药物与细菌的调控网络。

图1 药物成分-靶点可视化网络图Figure 1 Visual network diagram of drug components-targets
2.2 药物-细菌PPI网络分析
在Uniprot数据库中可找到有结构的Pg蛋白为1 865个。
268个亚甲基蓝潜在靶点和1 865个Pg的蛋白之间有19个共同的靶点,这19个靶点为:groS、radA、rplA、dps、fabH、pyrG、thyA、panC、RHO、frdA、ileS、bioA、def、ddl、TPR、murA、lepB、cobT、gyrB。
通过数据库String对亚甲基蓝靶点和Pg的靶点进行蛋白质相互作用,形成亚甲基蓝对Pg的调控网络(图2),空节点代表未知3D结构的蛋白,填充节点为已知或预测结构的蛋白,边(灰色线条)代表蛋白之间的关系,线颜色越深代表边的置信度越高。

图2 药物-细菌共同作用蛋白的PPI网络图Figure 2 PPI network diagram of drug-bacterial PPI diagram
本研究通过对PPI分析发现,一共有19个节点,涉及12个相互作用的边,8个孤立的节点(tpr、frdA、radA、dps、panC、lepB、def、cobT)暂未发现参与PPI,这些蛋白的功能或者未知,或者主要与核糖体或代谢相关,与Pg已知的常见致病蛋白关联不紧密,需要进一步分析筛选蛋白。
2.3 分子对接
经筛选并使用AutoDock 运算分析,得到亚甲基蓝与潜在结合蛋白受体的结合能为:FimA:-6.26 kcal/mol,Mfa4:-5.91 kcal/mol,RgpB:-5.14 kcal/mol,Kgp K1:-5.07 kcal/mol。
2.3.1亚甲基蓝与FimA蛋白受体结合的最佳复合结构 FimA蛋白是Pg主要菌毛的结构亚基,如图3所示,亚甲基蓝与FimA蛋白的 LEU51、ALA124、GLN127、ALA250、SER251、ASN253、THR254、GLY261、PHE262 这9个位点产生结合,其结合能为-6.26 kcal/mol。
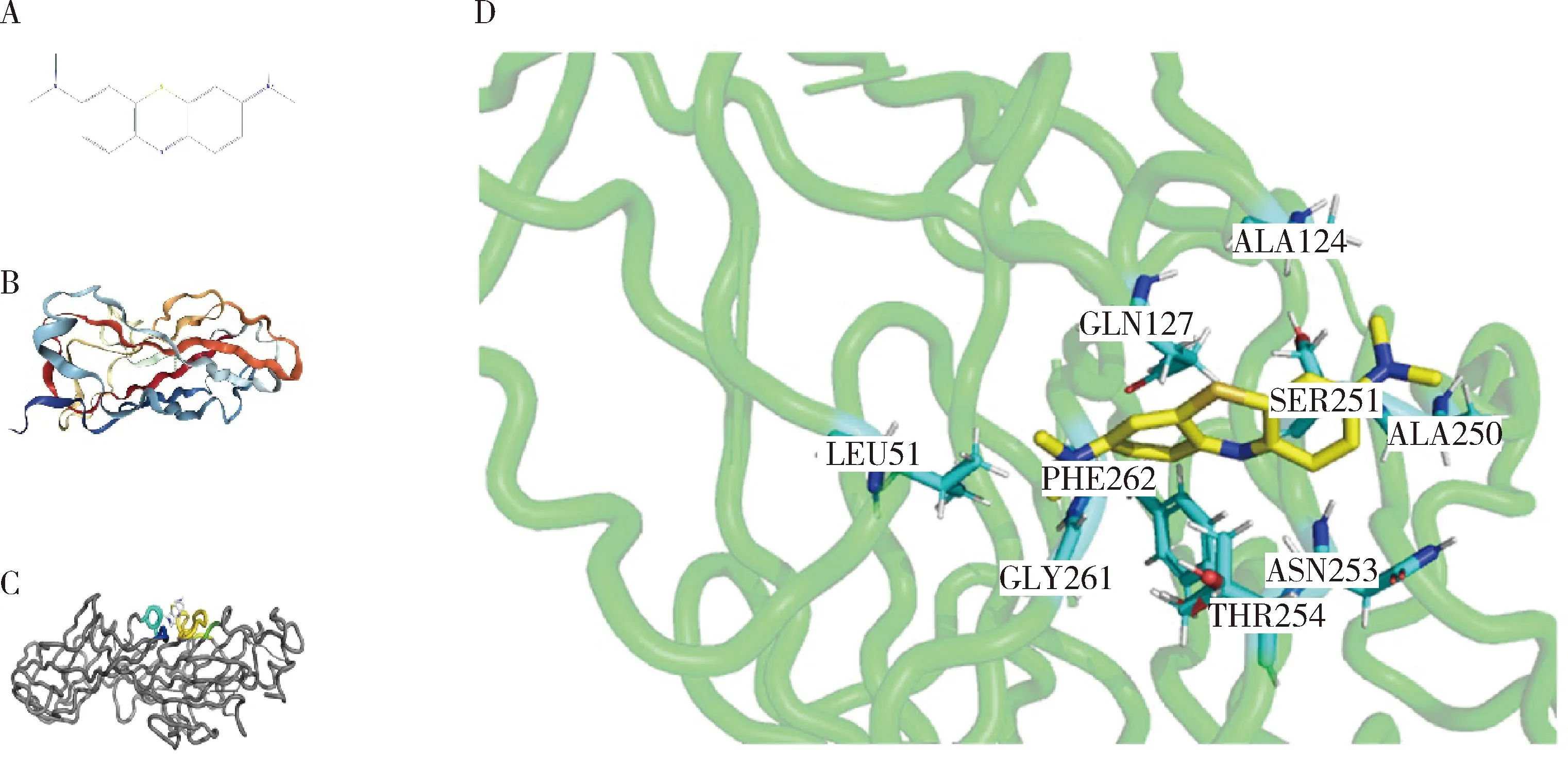
A, methylene blue structure; B, FimA protein structure; C, methylene blue and FimA protein docking (overall); D, methylene blue and FimA protein docking (partial).图3 亚甲基蓝与靶蛋白FimA的分子对接模式图Figure 3 Molecule docking diagram of methylene blue and target protein FimA
2.3.2亚甲基蓝与Mfa4蛋白受体结合的最佳复合结构 Mfa4蛋白是与大菌毛蛋白FimA相对应的小菌毛的尖端亚基,如图4所示,亚甲基蓝与Mfa4蛋白的GLY46、GLU330、LYS328、GLU52这4个位点产生结合,氢键形成位点为GLU47,结合能为-5.91 kcal/mol。
2.3.3亚甲基蓝与RgpB蛋白受体结合的最佳复合结构 RgpB蛋白催化FimA的成熟,还能水解蛋白质和小分子底物,如图5所示,亚甲基蓝与RgpB蛋白的氢键LYS80形成强结合位点,其结合能为-5.14 kcal/mol。

A, methylene blue structure; B, RgpB protein structure; C, methylene blue and RgpB protein docking (overall); D, methylene blue and RgpB protein docking (partial).图5 亚甲基蓝与RgpB蛋白的分子对接模式图Figure 5 Molecule docking model diagram of methylene blue and RgpB protein
KEYWORDSTarget prediction; Molecular docking; Photodynamic; Methylene blue;Porphyromonasgingivalis
2.3.4亚甲基蓝与Kgp K1蛋白受体结合的最佳复合结构 Kgp K1蛋白参与细菌的共聚集,辅助生物膜的形成,如图6所示,亚甲基蓝与Kgp K1蛋白的TYR1020、ALA1018、GLN1113、THR1115、TRP1116、TYR1020这6个位点产生结合,氢键形成位点GLY1114,结合能为-5.07 kcal/mol。

A, methylene blue structure; B, Kgp K1 protein structure; C, methylene blue and Kgp K1 protein docking (overall); D, methylene blue and Kgp K1 protein docking (partial).图6 亚甲基蓝与Kgp K1蛋白的分子对接模式图Figure 6 The molecular docking diagram of methylene blue and Kgp K1 protein
3 讨论
抗菌光动力疗法因其可以通过按需光照,控制药物杀菌范围、时间,降低细胞毒性,逐步受到研究人员的重视。
光动力杀菌的前提是光敏剂与细菌接触结合,光敏剂结合、内化到细菌中受光敏剂结构、疏水性程度、正电荷数量等影响[26],而细菌的结构又与致病机制密切相关。光敏剂如果能结合细菌的生物膜、毒力相关蛋白,则可以进一步解释光动力治疗的杀菌机制。
在已获得结构解析的Pg蛋白中,FimA蛋白是主要菌毛的结构亚基,促进附着在细菌表面长的丝状菌毛介导生物膜的形成,也与细菌的黏附功能相关,以及在牙周疾病发生、发展中发挥重要作用[27-28]。属于Mfa蛋白簇之一的Mfa4蛋白也可诱导蛋白参与菌毛的形成过程[27],其致病作用包括表面黏附、细胞间相互作用以及促进生物膜形成。RgpB通过添加阴离子多糖产生膜型RgpB,在细菌表面或外膜囊泡上的黏附和结合过程起主要作用。Kgp K1与血红蛋白有良好的结合作用,在生物膜的形成过程中促进了细菌的定植,增强Pg毒力[29]。然而,现有的抗菌光动力疗法的机制研究中,在光敏剂与细菌蛋白的结合作用机制的探索较少,其原因可能是缺少一个有效的工具来预测、筛选和初步验证光敏剂与细菌蛋白的结合程度。
本研究使用PharmMapper数据库、Uniprot数据库对光敏剂亚甲基蓝与牙龈卟啉单胞菌可能结合的蛋白靶点进行预测,使用Autodock分子对接软件进行分析。通过计算机反向模拟PharmMapper数据库的药效团模型,发现亚甲基蓝可以潜在地干预Pg的核苷酸的合成和代谢,进而抑制其增殖,但由于PharmMapper数据库药效团模型所收集的Pg蛋白靶点的特异性不高,无法完成亚甲基蓝对Pg蛋白靶点的精确定位,因此本研究利用AutoDcok软件将亚甲基蓝与Pg的FimA蛋白、Mfa4蛋白、RgpB蛋白、Kgp K1蛋白进行分子对接,寻找其靶向这些蛋白的活性中心和结合位点。
有研究认为,可将结合能作为判断高结合性的精确筛选标准,结合能小于0 kJ/mol则表明配体分子可以与受体分子自发结合。目前有很多报道,如尧晨光等[30]发现最低结合能为-0.99 kcal/mol时,配体与受体结合较好。刘福和等[31]发现最低结合能为-1.07 kcal/mol为高结合性分数。Tsvetkov等[23]检测则发现,光敏剂Ce6与两亲性聚合物配合物结合能在-4 kcal/mol左右,因此,本研究选用“结合能”作为筛选亚甲基蓝与细菌蛋白结合程度的指标,且本研究使用更高的标准,设置结合能≤ -5.0 kcal/mol为标准进行筛选。
通过从PDB数据库下载FimA、Mfa4、RgpB和Kgp K1蛋白靶点的三维结构文件,使用AutoDock软件将亚甲基蓝文件和这些靶点三维结构文件进行正向分子对接,计算各靶点结合自由能,结果显示亚甲基蓝可以与这么蛋白产生明显的结合能力,具体体现在其结合位点数目从1个到9个不等,结合能在-5.07~-6.26 kcal/mol(均小于-5 kcal/mol)。此外,本研究尝试了已解析出结构但未明确功能的PG1108蛋白,发现其结合能为-3.5 kcal/mol,其值大于-5 kcal/mol,在本研究中认为结合能较弱而未列出。
本研究结果表明,亚甲基蓝可以结合Pg的部分蛋白,光照时亚甲基蓝产生的活性氧可以破坏这些蛋白,导致细菌死亡。
综上,本研究基于靶点预测和分子对接的方式,研究光敏剂亚甲基蓝和Pg相关蛋白在光动力疗法中的结合情况,为研究光敏剂与生物大分子蛋白的结合位点和结合程度提供研究手段,可提高光敏剂作用于蛋白靶点的筛选效率,为进一步验证相关作用机制提供参考。
猜你喜欢
分子催化(2022年1期)2022-11-02
分子催化(2022年3期)2022-08-13
安徽医科大学学报(2022年4期)2022-05-12
世界中医药(2021年22期)2021-01-03
佛山陶瓷(2018年6期)2018-09-14
上海医药(2018年15期)2018-09-03
科技创新导报(2016年30期)2017-03-15
当代化工(2015年6期)2015-10-21
中学理科·综合版(2008年11期)2008-01-14
