
调控CuO表面性质选择性电催化还原CO2制HCOOH
2022-04-12 10:42郭李娜何雪冰吕琳吴丹原弘
无机材料学报 2022年1期
郭李娜, 何雪冰, 吕琳, 吴丹, 原弘
调控CuO表面性质选择性电催化还原CO2制HCOOH
郭李娜, 何雪冰, 吕琳, 吴丹, 原弘
(华中师范大学 化学学院, 农药与化学生物学教育部重点实验室, 武汉 430079)
电催化二氧化碳还原反应可将温室气体二氧化碳转化为化工原料或者有机燃料, 为克服全球变暖和电能向化学能转化提供了一条可行途径。该技术的主要挑战是产物分布广, 导致单一产物选择性低, 而调控催化剂的表面性质是解决这一难题的可行策略。本研究通过对氧化亚铜、硫化亚铜进行氧化制备表面性质不同的氧化铜, 其中, 氧化硫化亚铜制得的CuO-FS催化剂提高了电还原二氧化碳的活性和还原产物甲酸的选择性。该催化剂表现出较高的总电流密度, 而且在一个较大的测试电压范围(–0.8 ~ –1.1 V)内, 甲酸的法拉第效率可以保持在70%以上, 在–0.9 V时达到最大值78.4%。反应机理探究表明, CuO-FS优异的电还原二氧化碳性能归因于其较大的电化学活性表面积提供了大量表面活性位点, 产生较高的总电流密度; 而且电催化过程中催化剂表面产生较少的零价Cu, 减少了乙烯的生成, 使产物更集中于甲酸。
电还原二氧化碳; 铜基催化剂; 表面性质; 产物选择性; 甲酸
近年来, 煤、石油、天然气等化石能源的大量燃烧导致二氧化碳(CO2)排放量日益增长, 从而引起温室效应、全球变暖等环境问题[1-2]。因此, 开发新型的清洁能源以及实现CO2的减排已经迫在眉睫。自然界中, 植物可以通过光合作用将CO2和水(H2O)转化为有机物和氧气(O2), 受此启发, 研究人员将目光转向了CO2还原这一课题上。将CO2还原为高附加值的化工原料或燃料是克服全球变暖和能源供应短缺问题的有效战略之一[3], 同时也符合“碳中和”的理念。目前, 实现CO2还原主要有电催化[4]、光催化/光热催化[5-6]和热催化[7]等多种技术, 但上述技术仍然存在能量转化效率低等问题。
相较于其他技术, 电催化CO2还原(CO2reduction reaction, CO2RR)具有温和、环境友好、能量转换效率高等优点[8]。然而, 电催化反应是在水溶液中进行的氧化还原过程, 析氢反应(Hydrogen evolution reaction, HER)是CO2RR的主要竞争反应, 因此抑制HER成为电催化反应的一项巨大挑战[9]。除此之外, CO2RR反应产物的还原电势接近[10], 导致产物分布较广(包括一氧化碳(CO)[11]、甲酸(HCOOH)[12]、甲烷(CH4)[13]、甲醇(CH3OH)[14]、乙烯(C2H4)[15]、乙酸(CH3COOH)[16]、乙醇(C2H5OH)[17]等)。其中, HCOOH是一种应用广泛的工业原料, 可以直接用于生产消毒剂和防腐剂以及合成甲酸盐、甲酸酯。近年来, 大量研究发现锡(Sn)、铅(Pb)、铟(In)及铋(Bi)基[18-21]等电催化剂可以将CO2催化还原为HCOOH。但是, 这些过渡金属的成本较高且HCOOH选择性低, 因此仍亟待开发低成本、高HCOOH选择性的替代材料。铜(Cu)基催化剂以其原料来源广泛、价格低廉且性能优异等优点, 在诸多过渡金属基催化剂中脱颖而出, 受到了广泛关注; 它是唯一可以促进CO2RR发生碳碳键(C–C)偶联制备C2H4的催化剂, 对制备高价值的多碳产物具有重要的研究意义[22]。但是Cu基催化剂的表面特性较为敏感, 产物分布广泛, 产物选择性较难控制。因此, 通过对Cu基催化剂改性将CO2高选择性地还原为HCOOH也是该领域的一大难点。
对此, 本课题组构建了一种新型的钾铜双金属硫化物/氧化铜(KCu7S4/CuO)界面结构, 可诱导形成局部电荷区, 优化活性中间体在界面上的吸附特性, 从而实现CO2RR高选择性地制备HCOOH[23]。因此, 通过构建催化剂的表/界面可以对CO2RR反应产物的选择性进行调控。本研究通过简单的湿化学法合成了球状的氧化亚铜(Cu2O)和硫化亚铜(Cu2S), 并且进一步氧化生成两种纳米片状的氧化铜(CuO-FO, FO: from oxide; CuO-FS, FS: from sulfide)。其中, CuO-FS表现出优异的CO2RR性能, 即较大的总电流密度和较集中的产物分布, 其在较低的过电位下产生较高的HCOOH法拉第效率(FEHCOOH), 并且有效地抑制了HER。此外, 本研究通过N2吸附/脱附、粉末X射线衍射及电子顺磁共振等表征技术分析了CuO-FS性能明显提升的原因。
1 实验方法
1.1 材料的制备
Cu2O的合成。称取483.2 mg三水合硝酸铜(Cu(NO3)2·3H2O), 加入25 mL乙二醇, 搅拌使固体完全溶解, 生成蓝色溶液。再加入0.15 g聚乙烯吡咯烷酮(Polyvinyl pyrrolidone, PVP), 搅拌10 min后将混合液转移至圆底烧瓶中, 油浴加热至180 ℃保温30 min, 制得黄褐色悬浊液。将其转移至离心管中, 用蒸馏水(H2O)和C2H5OH交替洗涤离心三次(离心速度: 10000 r/min, 离心时间: 5 min)。将所得黄褐色沉淀置于60 ℃烘箱中过夜直至干燥, 最终制得黄褐色氧化亚铜(Cu2O)粉末。
CuO-FO的合成。称取50 mg制备的Cu2O粉末, 加入20 mL氢氧化钾(KOH, 0.1 mol/L)溶液, 搅拌10 min使Cu2O粉末分散完全。再加入2 mL的双氧水(H2O2, 质量分数30%), 搅拌20 min后将其转移至反应釜中, 并置于烘箱中120 ℃加热24 h, 所得黑色沉淀分别用H2O和C2H5OH洗涤三次, 再置于60 ℃烘箱中过夜直至干燥得到最终产品。该样品由Cu2O氧化制得, 命名为CuO-FO。
Cu2S的合成。称取483.2 mg Cu(NO3)2·3H2O, 加入25 mL乙二醇, 搅拌使固体完全溶解, 生成蓝色溶液。再加入0.15 g PVP, 搅拌10 min后将混合液倒入圆底烧瓶中, 油浴加热至180 ℃保温30 min, 制得黄褐色悬浊液。将其快速转移至50 mL反应釜中, 并加入456.7 mg 硫脲(CH4N2S), 搅拌2 min, 将反应釜置于烘箱中180 ℃加热, 24 h后所得黑色沉淀分别用H2O和C2H5OH洗涤三次, 再置于60 ℃烘箱中过夜直至干燥, 制得黑色的硫化亚铜(Cu2S)粉末。
CuO-FS的合成。称取50 mg制得的Cu2S粉末, 加入20 mL KOH溶液(0.1 mol/L), 搅拌10 min使Cu2S粉末分散完全。再加入2 mL的H2O2(质量分数30%), 搅拌20 min后将其转移至反应釜中, 并置于烘箱中120 ℃加热, 24 h后所得黑色沉淀分别用H2O和C2H5OH洗涤三次, 置于60 ℃烘箱中过夜直至干燥得到最终产品。该样品由Cu2S氧化制得, 将其命名为CuO-FS。
1.2 材料表征
使用场发射扫描电子显微镜(Field emission scanning electron microscope, FESEM, Hitachi SU8010)观察样品的微观形貌; 使用透射电子显微镜(Transmission electron microscopy, TEM, FEI Tecnai G2 F30)观察样品的微观形貌, 并采用能量色散X射线谱(Energy dispersive spectroscope, EDS)分析元素分布等; 使用X射线衍射(X-ray diffraction, XRD, Bruker D8, 铜靶(0.15418 nm), 工作电压40 kV, 工作电流40 mA, )分析样品的晶体结构; 使用X射线光电子能谱(X-ray photoelectron spectroscopy, XPS, ESCALAB 250Xi, 铝靶(1486.6 eV), 灯丝电压14.7 keV、灯丝电流10 mA)分析样品中元素的化学价态; 使用电子顺磁共振(Electron paramagnetic resonance, EPR, Bruker A300)分析样品的表面缺陷。
1.3 电化学表征
所有电化学测量均在与CHI 760E电化学工作站(上海辰华)连接的H型电解池中进行, 电解池中使用全氟磺酸质子交换膜(Nafion 117)分隔两个反应室, 分别将铂(Pt)片电极和氯化银(Ag/AgCl)电极作为对电极和参比电极, 将1.0 cm×1.0 cm涂有催化剂(负载质量约为1.0 mg)的碳纸作为工作电极。工作电极的制备方法: 称取10 mg样品粉末溶于980 μL C2H5OH溶液和20 μL 全氟磺酸型聚合物(Nafion, 5.0wt%)溶液的混合液中, 超声40 min。用移液枪取100 μL 混合液滴涂于1 cm2碳纸上, 并使用红外灯照射进行干燥。通过称量滴涂前后碳纸质量的变化, 确认滴涂催化剂的质量约为1 mg。将碳酸氢钾(KHCO3, 0.1 mol/L)溶液用作电解液(每个反应室填充60 mL电解质溶液, 剩余40 mL顶部空间)。测试前, 将20 mL·min–1的CO2或氩气(Ar)流连续通入电解液中至少半小时, 并在整个电解过程中保持这种状态。
所有的极化曲线均以5 mV·s–1的扫描速率进行采集。通过在各种恒定电压下进行电化学反应来获得特定电压下的产物组成。通过计算电化学双层电容值来评估电化学活性表面积(Electrochemical activesurface area, ECSA), 计算公式为dl= Δ/2, 其中dl, Δ和分别是催化剂的电化学双电层电容(μF·cm–2), 电流密度差(mA·cm–2)和扫描速率(mV·s–1)。这项工作中的所有电压都用以下公式来校准:RHE=Ag/AgCl+ 0.059 × pH + 0.197 V。
1.4 产物分析
电化学反应生成的气态产物、液态产物(HCOOH)分别通过在线气相色谱仪(Gas chromatograph, GC; GC2030plus, 武汉泰特泰克科技有限公司)和核磁共振波谱仪(Nuclear magnetic resonance, NMR; VNMR600, 武汉中科牛津波谱技术有限公司)进行测试。测试前分别用标准气体和HCOOH标准样品对上述两个仪器进行校准, 制作标准曲线, 后续的产物浓度通过该标准曲线计算获得。在给定电压下, 气态产物和HCOOH的法拉第效率(FEgas和FEHCOOH; Faraday efficiency, FE)根据分别公式(1, 2)计算:
FEgas= (H×1×) /×100% = (H××××60×)/
(total×××)×100% (1)
其中, gas对应于H2、一氧化碳(CO)和C2H4三种气体;H: 形成气体产物的转移电子数, 对于H2和CO均为2, 而对于C2H4为12,: 法拉第常数, 96485 C·mol–1;: 环境压强, 1.013×105Pa;: 数字流量控制器在环境压力下测量的气体流速, 20 mL·min–1;: 从阴极室产生气体的体积浓度(体积百分比);total: 测得的稳态电流;: 时间, 60 s;: 气体摩尔常数, 8.314 J·mol–1·K–1;: 开尔文温度, 298.15 K;1: 气体产物的摩尔数;: 采样期间通过系统的总电量。
FEHCOOH= (F×2×)/×100% (2)
其中,F: 形成HCOOH的转移电子数, 其数值为2;2: HCOOH产物的摩尔数;: 法拉第常数, 96485 C·mol–1;: 采样期间通过系统的总电量。
通过仪器测量不同产物的峰面积, 利用标准曲线校正为浓度, 分别代入上述公式, 即可求出相应产物的法拉第效率。
2 结果与讨论
2.1 催化剂形貌和组分表征
用FESEM和TEM观察样品的微观形貌。从Cu2O和Cu2S的FESEM照片(图1(a, b))中可以看出, 两种前驱体微观形貌都是尺度为几十到几百纳米的球状颗粒。两者用H2O2氧化后, 制得的CuO-FO和CuO-FS都呈纳米片结构(图1(c, d)), 且两种纳米片的厚度及长度均相近。同样, 从CuO-FS的TEM照片中(图1(e))可以更加清楚地看到CuO的纳米片结构。通过高角环形暗场图像(图1(f))及其对应的EDS元素分布图(图1(g, h))对CuO-FS的形貌与组分进一步确认, 即CuO-FS是包含Cu、O两种元素的纳米片。总之, Cu2O和Cu2S是尺度为几十到几百纳米的球状颗粒, 氧化后所得的CuO-FO和CuO-FS是尺寸相近的纳米片, 两者的微观形貌无较大差别。

图1 样品的形貌与元素组成
FESEM images of (a) Cu2O, (b) Cu2S, (c) CuO-FO and (d) CuO-FS; (e) TEM image of CuO-FS; (f) HAADF image of the CuO-FS; (g, h) Corresponding EDS elemental mapping
对Cu2O、CuO-FO、Cu2S、CuO-FS四种催化剂分别进行了粉末XRD测试(图2), 通过与粉末衍射文件数据库(Powder diffraction file, PDF)的标准卡片比对, 可以确定四种样品的晶体结构。由Cu(NO3)2·3H2O回流加热制备得到黄褐色的Cu2O(JCPDS 77-0199), 加入CH4N2S后水热合成制备得到黑色的Cu2S(JCPDS 3-1071), XRD谱图显示这两种前驱体均不存在其他杂质。制得Cu2O和Cu2S后, 对二者分别进行氧化处理, 最终制得的两种黑色粉末均为CuO(JCPDS 80-1916)。因此可以确定: 氧化生成的两种样品CuO-FO与CuO-FS在晶体结构上无较大差别。
为了进一步研究CuO-FO和CuO-FS样品中元素的化学价态, 对这两种样品进行XPS测试(图3(a, b))。XPS全谱测试显示: CuO-FO和CuO-FS都只含有Cu和O两种元素, 这确定了二者组分的相似性。为了进一步比较二者的差别, 对这两种元素进行细化分析(图3(c, d)), Cu2p轨道的高分辨XPS谱图显示: CuO-FS与CuO-FO的峰数量和峰位置无明显差别, 均在953.4 eV处出现Cu2p1/2信号峰及其相应卫星峰, 且在933.6 eV处出现Cu2p3/2信号峰及其相应卫星峰。这两种信号峰均与+2价的Cu相关, 符合XRD的测试结果, 再次证明CuO-FO与CuO-FS两种样品均为CuO。但是, 这两者的O1s轨道的高分辨XPS谱图又存在差别。O1s轨道的XPS信号峰主要分为晶格氧的峰(Olatt)和表面氧的吸附峰 (Oads), 而Oads又可分为水蒸气(H2O)的吸附峰(OH)以及CO2的吸附峰(OC)。两者的Olatt近乎一致, 而相比CuO-FS, CuO-FO的OH和OC峰占比增加, 由于XPS是在真空条件下测试的, 所以这表明CuO-FO对反应物H2O和CO2的化学吸附性比CuO-FS更强。

图2 Cu2O、Cu2S、CuO-FS和CuO-FO的XRD图谱
2.2 电化学性能
将一个三电极的H型电解池(对电极为Pt片电极, 参比电极为Ag/AgCl电极)与CHI 760E电化学工作站相连, 注入CO2饱和的0.1 mol/L KHCO3溶液作为电解液来测试催化剂的CO2RR活性。首先, 在0~–1.2 V(RHE)测试电压范围内, 以50 mV·s–1速率扫描, 通过连续循环伏安法(Cyclic voltammetry, CV)测试激活样品, 获得稳定的CV曲线后, 在同一测试电压, 对CuO-FO和CuO-FS进行线性扫描伏安法(Linear sweep voltammetry, LSV)测试。
阴极极化曲线(图4(a))显示: 与注入Ar饱和的KHCO3溶液中的电流密度相比, CuO-FO和CuO-FS在注入CO2饱和的KHCO3溶液中检测到的电流密度都有增加, 表明两者均有CO2RR催化活性。而且, 几乎在整个测试电压范围内, CuO-FS的CO2RR活性都更高, 尤其是随着电压减小, 其两条极化曲线的差距逐渐加大, 这表明CuO-FS上的HER逐渐被抑制, 电子更倾向于运输到CO2还原的活性位点上用于产生HCOOH。与CuO-FS相比, CuO-FO上两条极化曲线的差距较小, 尤其是在高电压处, 对HER的抑制作用较小。图4(b~e)分别显示Cu2S、Cu2O、CuO-FO和CuO-FS四个样品的CO2RR总电流密度、产物组成及相应的法拉第效率。从总电流密度看, Cu2S、Cu2O和CuO-FS三个样品在各测试电压下均明显优于CuO-FO。从产物选择性看, Cu2S的产物组成不同于其他三者, 只产生H2和HCOOH, 而且FEH2高达约80%, 这表明电催化过程中多数电子参与了HER反应。然而, Cu2O、CuO-FO和CuO-FS作为催化剂都有四种产物, 分别为HCOOH、H2、CO和C2H4。但这三者中, CuO-FS的产物选择性最高, 主要产生HCOOH, 在–0.9 V时FEHCOOH高达78.4%, Cu2O和CuO-FO在同等测试电压下的FEHCOOH仅为53.5%和44.4%。而且, 在一个较大的测试电压范围(–0.8 ~ –1.1 V)内(图4(f)), CuO-FS的FEHCOOH可以保持在70%以上(具体为: –0.8 V, 71.5%; –0.9 V, 78.4%; –1.0 V, 76.3%; –1.1 V, 73.4%); 随着电压减小, 其FEH2不断减少, 这表明在电催化过程中CuO-FS有效地抑制了HER, 这与CuO-FS的阴极极化曲线相对应。此外, 这四种样品中, CuO-FO的FEC2H4最高, 在–1.2 V时可达23.1%, 这也验证了文献[24]的结论: Cu基催化剂易发生C–C偶联生成C2H4。综上, CuO-FO和CuO-FS的微观形貌与晶体结构无明显差异, 但两者的CO2RR性能差别显著, CuO-FO产生C2H4较多, 而CuO-FS可抑制CO2RR发生C–C偶联, 高选择性地生成HCOOH。这充分证明: 调控催化剂的表面性质即可调控电催化CO2RR性能。
如图5(a, b), 在Cu2S、Cu2O、CuO-FO和CuO-FS四种样品中, CuO-FS的FEHCOOH及HCOOH部分电流密度在整个测试电压范围内都居于最高位置; 其FEHCOOH在–0.9 V时高达78.4%, 且HCOOH的部分电流密度在–1.2 V时高达11.9 mA·cm–1。因此, CuO-FS在CO2还原为HCOOH的过程中有着显著的优势。为了进一步探究电催化CO2还原为HCOOH的动力学过程, 我们从电压和电流密度的相关性中转换得到Tafel曲线(电流密度的对数与过电位的关系), Tafel斜率可以用来表示电催化反应过程中的相对内阻, 一种性能优异的电化学催化剂应具有较低的Tafel斜率和较大的电流密度[25]。如图5(c), CuO-FS的Tafel斜率(263.3 mV·dec–1)远小于CuO-FO(309.8 mV·dec–1), 这表明两种催化剂的电化学催化机理不同, 同时也证明改变CuO的表面性质可大大减少CO2RR的动力学阻碍。此外, 催化活性位点的数量与催化活性和产物选择性高度相关。通过测试双层电容可以确定催化剂的ECSA。首先, 测试CuO-FO和CuO-FS在不同测试电压扫描速度(10~60 mV·s–1)下的CV曲线(图5(d, e)), 再利用1.3节公式计算其对应的双电层电容(图5(f))。结果显示, CuO-FS的双电层电容(7.1 μF·cm–2)约为CuO-FO(3.9 μF·cm–2)的两倍, 这证实了调控CuO的表面性质可提高活性位点的数量。
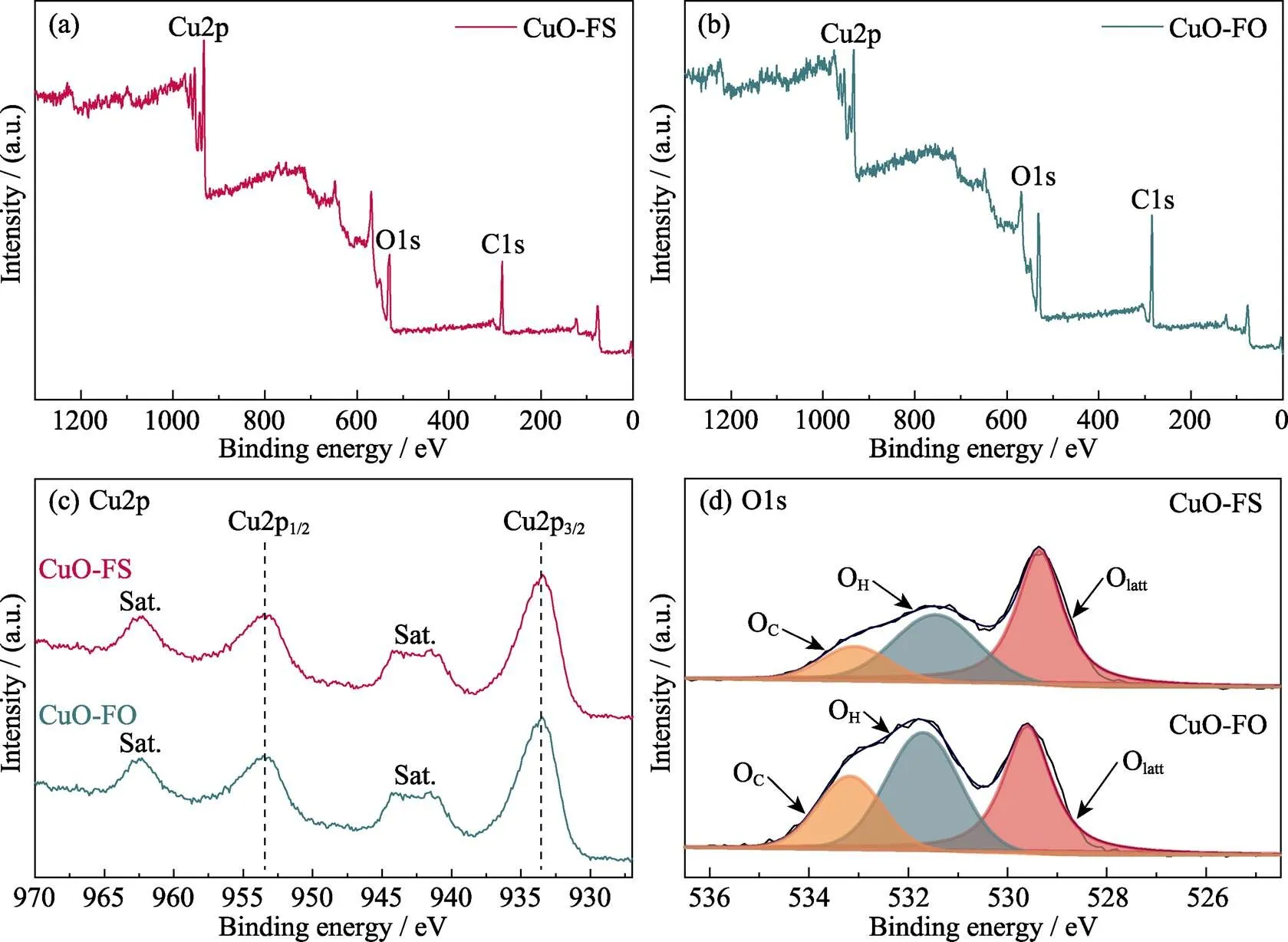
图3 CuO-FS和CuO-FO的XPS图谱
(a, b) Survey spectrum; (c) High resolution XPS spectra of Cu2p; (d) High resolution XPS spectra of O1s

图4 (a)CuO-FO和CuO-FS在注入CO2/Ar饱和的0.1 mol/L KHCO3电解液中的阴极极化曲线; (b)Cu2S、(c)Cu2O、(d)CuO-FO和 (e)CuO-FS上所有产物的法拉第效率和电流密度; (f)CuO-FS在不同测试电压下所有产物的法拉第效率
Colorful figures are available on website
Cu基催化剂较差的稳定性一直阻碍着CO2RR的发展[26], 为测试CuO-FS在电催化过程中的稳定性, 在–0.9 V电压下对其进行长达20 h的稳定性测试(图6), 结果显示: CuO-FS的电流密度在20 h内未发生衰退, 其FEHCOOH平均值为74.5%, 这表明CuO-FS对电还原CO2制备HCOOH有良好的稳定性。
2.3 CuO-FS电化学还原性能提升的原因
根据前文讨论, CuO-FO和CuO-FS的微观形貌相似且晶体结构无明显差异, 但其CO2RR的性能差别显著: 在CO2RR过程中, CuO-FS的总电流密度高于CuO-FO; 在产物选择性方面, CuO-FO倾向于生成C2H4, 而CuO-FS可抑制C–C偶联, 从而提高HCOOH的选择性。为了探明CuO-FS电化学还原性能提升的原因, 对催化剂的表面性质开展研究。首先, 测试了CuO-FO和CuO-FS的N2吸附/脱附曲线, 并通过BET方程计算出样品的比表面积和孔体积。CuO-FO和CuO-FS的N2吸附等温曲线(图7)显示两者均有明显的迟滞回线, 这表明CuO-FO和CuO-FS都具有孔道结构。此外, CuO-FS的BET比表面积和孔体积(311.66 m2·g–1和0.5552 cm3·g–1)均明显高于CuO-FO(221.16 m2·g–1和0.4957 cm3·g–1), 表明CuO-FS的物理吸附能力高于CuO-FO, 这有利于CO2在催化剂表面的吸附。而且CO2在初始阶段能够吸附于催化剂表面是电催化CO2RR的先决条件, 较强的CO2吸附可以加快后续反应的动力学速率[27]。因此, 增大催化剂的比表面积和孔体积, 有利于提供更多的CO2还原活性位点, 这一结论与双电层电容的测试结果一致。尽管XPS结果显示CuO-FO样品对于反应物具有更强的化学吸附, 但是与该结果相比, 反应物在CuO催化剂上的物理吸附对电催化CO2RR性能的影响更大, 这是CuO-FS催化剂比CuO-FO催化剂具有更大电流密度的根本原因。
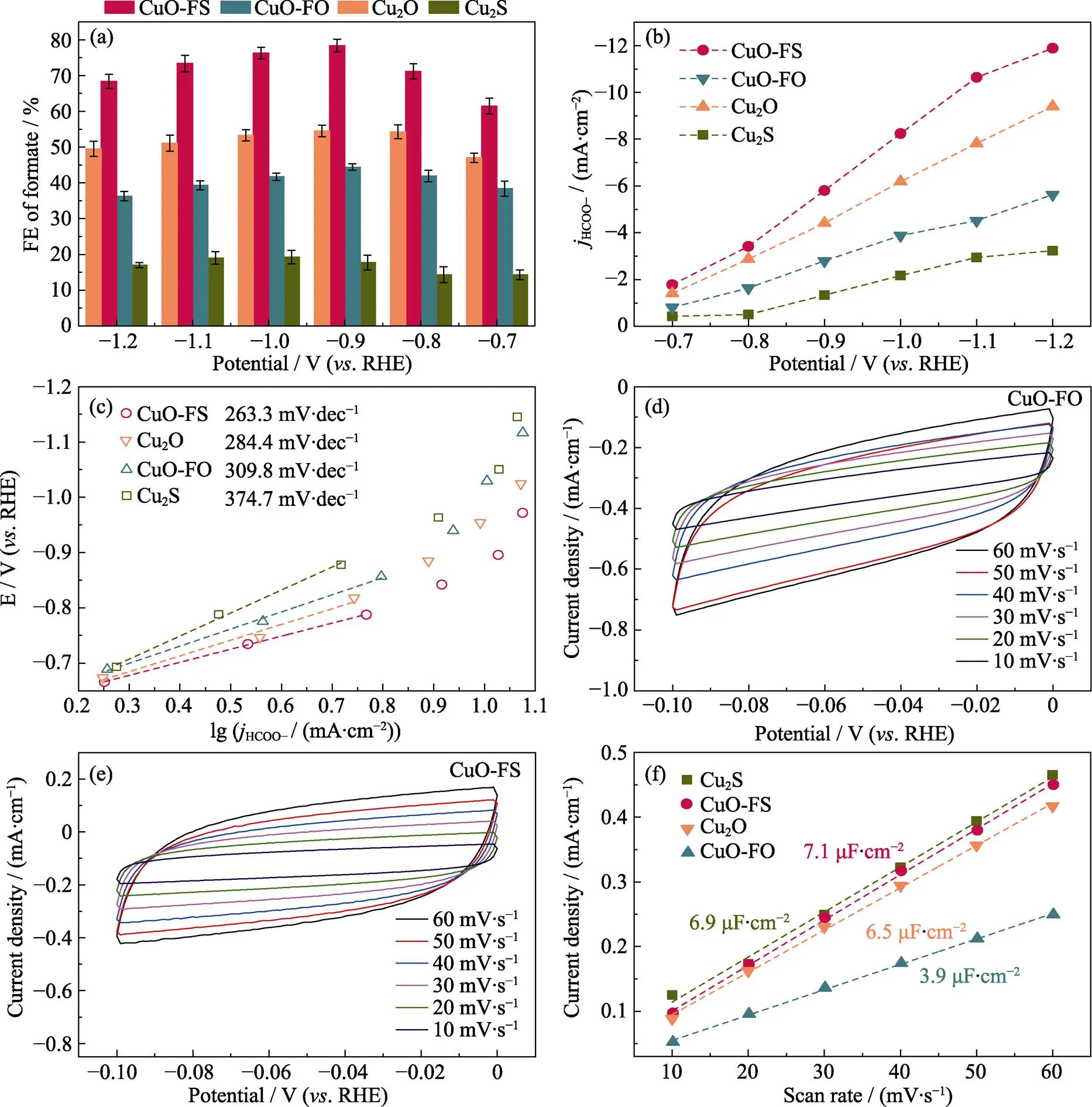
图5 Cu2S、Cu2O、CuO-FO和CuO-FS上HCOOH的 (a) 法拉第效率, (b) 部分电流密度和(c) Tafel曲线; (d) CuO-FS和 (e) CuO-FO在不同测试电压扫描速率下的CV曲线; (f) 四种样品在不同测试电压扫描速率的电流密度
Colorful figures are available on website
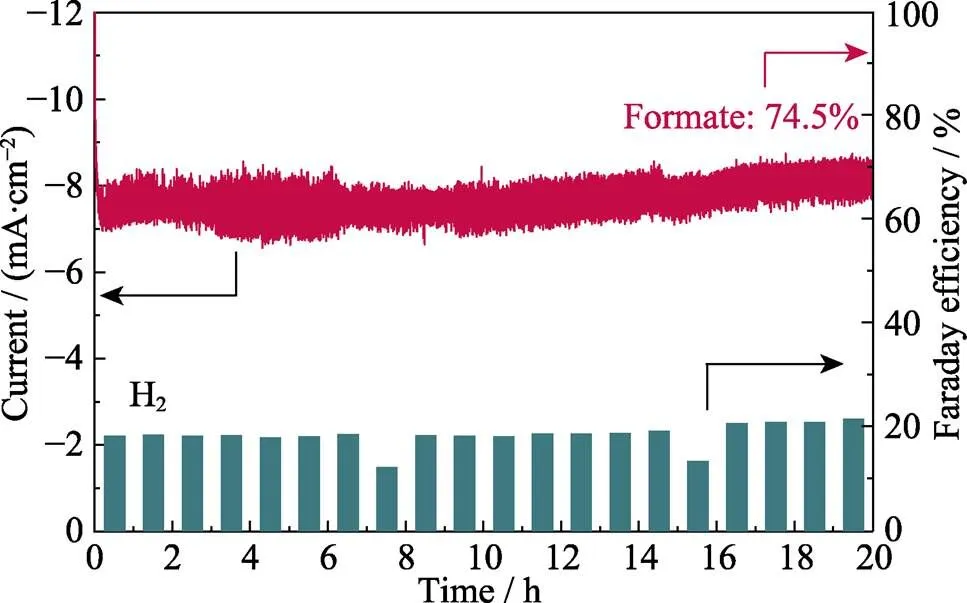
图6 CuO-FS在–0.9 V下的长期稳定性测试以及不同产物对应的法拉第效率
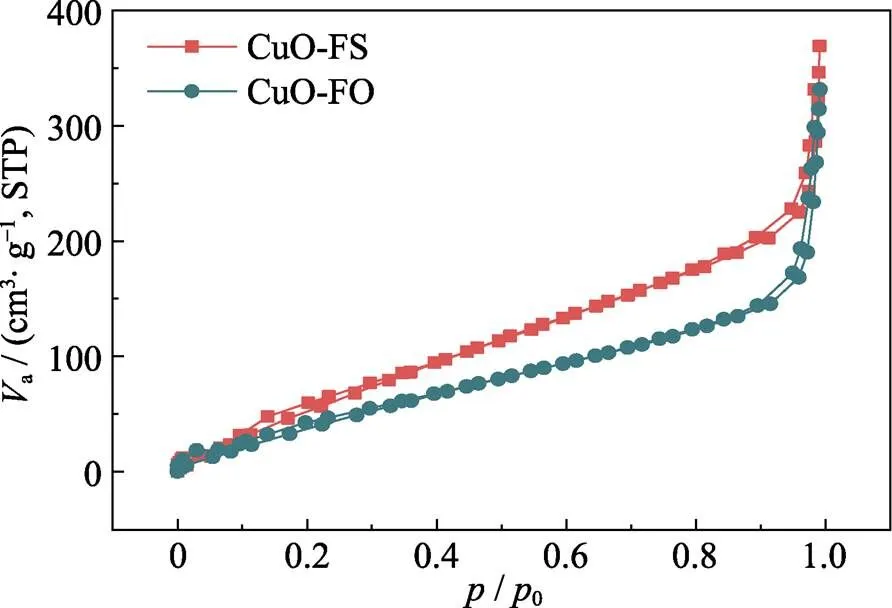
图7 CuO-FS和CuO-FO的N2吸附等温曲线
其次, Cu基催化剂中Cu的氧化物种类(对应Cu的不同价态)对产物的选择性具有重要影响[28], 因此, 实验对不同反应阶段(3 min和20 h)的CuO-FO和CuO-FS进行了XRD测试(图8(a, b))。结果显示: 在电催化CO2RR过程中, 两者都会产生Cu2O(含+1价Cu)和金属Cu(含零价Cu)的混合态; 而且在反应3 min和20 h后, CuO-FO中的零价Cu含量都高于CuO-FS。根据前人研究[28], +1价Cu和零价Cu的混合价态会同时促进两种反应中间体CObridge(bridge-adsorbed CO)和COatop(atop-adsorbed CO)的产生, 这是提高C2H4选择性的重要条件。
在本研究的CO2RR过程中, CuO-FO中零价Cu的出现较早且多, 造成其具有较高的C2H4法拉第效率。此外, 为了进一步探明电化学测试后两种催化剂表面Cu物种价态的变化情况, 实验对电化学测试1 h后的催化剂进行了EPR测试(图9)。结果显示: 两种催化剂均在= 2.00处出现EPR信号峰, 表明两者均存在O缺陷[29], 这种O缺陷是由+2价Cu还原为+1价Cu及零价Cu引起的。而且CuO-FO的O缺陷信号较CuO-FS强, 表明电化学测试1 h后, CuO-FO产生了较多的+1价Cu与零价Cu, 造成其具有较高的C2H4法拉第效率, 这与XRD结果相一致。综上, CuO-FS在CO2RR过程中产物选择性高是由于在电催化反应过程中零价Cu的成分较少, 减少了C2H4的生成, 从而提高了HCOOH的选择性。
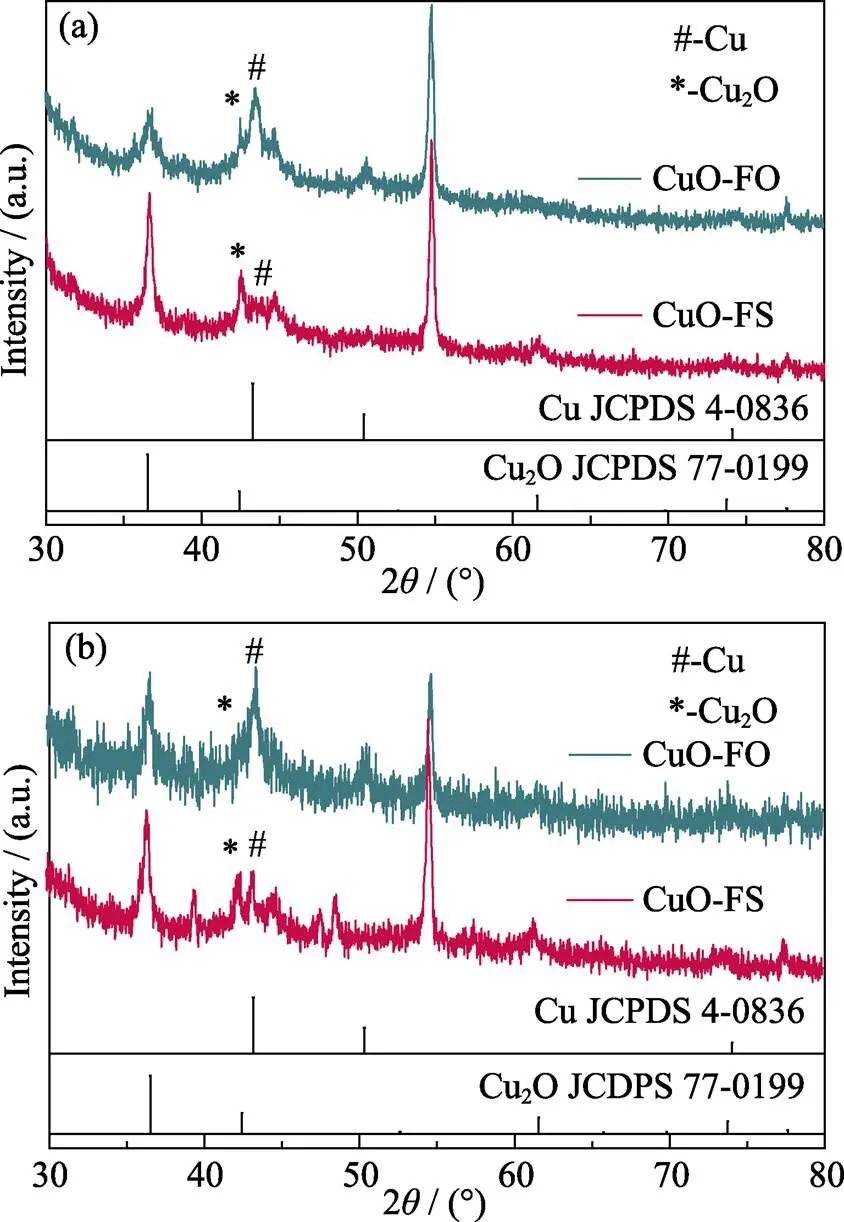
图8 CuO-FS及CuO-FO进行 (a) 3 min和 (b) 20 h测试后的XRD图谱

图9 电化学测试1 h后的CuO-FS及CuO-FO的EPR谱图
3 结论
本研究通过简单的湿化学法合成了球状的Cu2O和Cu2S, 并且进一步氧化生成两种纳米片状的CuO-FO和CuO-FS催化剂, CuO-FS表现出较优的CO2RR性能, 即较大的总电流密度和较集中的产物分布。该催化剂的FEHCOOH在–0.9 V时达到最大值78.4%, 电流密度为7.4 mA·cm–2; 且在一个较大的测试电压范围(–0.8~–1.1 V)内, FEHCOOH可以保持在70%以上; FEH2则随电压增加而不断减小, HER被有效地抑制。
通过N2吸附/脱附及粉末XRD等表征技术分析CuO-FS 催化剂具有较优CO2RR性能的原因如下: (1)较大的电化学活性表面积提供了大量表面CO2还原活性位点, 使CuO-FS比CuO-FO具有更大的总电流密度; (2)电催化过程中, CuO-FS催化剂产生零价Cu较少, 减少了C2H4的生成, 使其产物更集中于HCOOH。本研究通过调控催化剂的表面性质提升了电催化CO2RR反应的性能, 对影响Cu基催化剂选择性的因素进行了探讨, 这对电催化CO2还原反应以及电催化材料的研究提供了有益借鉴。
[1] VASILEFF A, YAO Z, SHI Z Q. Carbon solving carbon's problems: recent progress of nanostructured carbon-based catalysts for the electrochemical reduction of CO2., 2017, 7(21): 724-761.
[2] PETERS, GLEN, ANDERSON,. The trouble with negative emissions., 2016, 354(6309): 182–183.
[3] ZHANG S, ZHAO S, QU D,. Electrochemical reduction of CO2toward C2valuables on Cu@Ag core-shell tandem catalyst with tunable shell thickness., 2021, 2102293.
[4] XU K, NING S, CHEN H,. Plum pudding-like electrocatalyst of N-doped SnO@Sn loaded on carbon matrix to construct photovoltaic CO2reduction system with solar-to-fuel efficiency of 11.3%., 2020, 4(7): 2000116.
[5] QI Y, SONG L, OUYANG S,. Photoinduced defect engineering: enhanced photothermal catalytic performance of 2D black In2O(3–x)nanosheets with bifunctional oxygen vacancies., 2020, 32(6): 1903915.
[6] LI R, LI Y, LI Z,. A metal-segregation approach to generate CoMn alloy for enhanced photothermal conversion of syngas to light olefins., 2020, 5(2): 2000488.
[7] ZHANG C, CAO C, ZHANG Y,. Unraveling the role of zinc on bimetallic Fe5C2-ZnO catalysts for highly selective carbon dioxide hydrogenation to high carbon α-olefins., 2021, 11(4): 2121–2133.
[8] PADILLA M A, LU Q, BATURINA O A. CO2electroreduction to hydrocarbons on carbon-supported Cu nanoparticles., 2014, 4(10): 3682–3695.
[9] DUAN X, XU J, WEI Z,. Metal-free carbon materials for CO2electrochemical reduction., 2017, 29: 1701784.
[10] JIN S, HAO Z, ZHANG K,. Advances and challenges for electrochemical reduction of CO2to CO: from fundamental to industrialization., 2021, 60: 2–24.
[11] GU J, HSU C S, BAI L,. Atomically dispersed Fe3+sites catalyze efficient CO2electroreduction to CO., 2019, 364(6445): 1091–1094.
[12] LIU G, LI Z, SHI J,. Black reduced porous SnO2nanosheets for CO2electroreduction with high formate selectivity and low overpotential., 2019, 260: 118–134.
[13] LIN L, LIU T, XIAO J,. Enhancing CO2electroreduction to methane with cobalt phthalocyanine and zinc-nitrogen-carbon tandem catalyst., 2020, 59(50): 22408–22413.
[14] YANG D, ZHU Q, CHEN C,. Selective electroreduction of carbon dioxide to methanol on copper selenide nanocatalysts., 2019, 10(1): 1–9.
[15] DINH C T, BURDYNY T, KIBRIA M,. CO2electroreduction to ethylenehydroxide-mediated copper catalysis at an abrupt interface., 2018, 360(6390): 783–787.
[16] ZANG D, LI Q, DAI G,. Interface engineering of Mo8/Cu heterostructures toward highly selective electrochemical reduction of carbon dioxide into acetate., 2020, 281: 119426.
[17] LV X, SHANG L, ZHOU S,. Electron-deficient Cu sites on Cu3Ag1catalyst promoting CO2electroreduction to alcohols., 2020, 10 (37): 2001987.
[18] ZU X, LI X, WEI L,. Efficient and robust carbon dioxide electroreduction enabled by atomically dispersed Sn+sites., 2019, 31(15): 1808135.
[19] SHI Y, JI Y, LONG J,. Unveiling hydrocerussite as an electrochemically stable active phase for efficient carbon dioxide electroreduction to formate., 2020, 11(1): 3415.
[20] ZHANG A, LIANG Y, LI H,surface reconstruction of InN nanosheets for efficient CO2electroreduction into formate., 2020, 20(11): 8229–8235.
[21] SUN J, ZHENG W, LYU S,Bi/Bi2O3nanoparticles supported on N-doped reduced graphene oxide for highly efficient CO2electroreduction to formate., 31(6): 8229–8235.
[22] NITOPI S, BERTHEUSSEN E, SCOTT S B,. Progress and perspectives of electrochemical CO2reduction on copper in aqueous electrolyte., 2019, 119(12): 7610−7672.
[23] LV L, HE X, WANG J,. Charge localization to optimize reactant adsorption on KCu7S4/CuO interfacial structure toward selective CO2electroreduction., 2021, 298: 120531.
[24] XIE H, WANG T, LIANG J,. Cu-based nanocatalysts for electrochemical reduction of CO2., 2018, 21: 41–54.
[25] WANG X, WANG Z, ZHUANG T T,. Efficient upgrading of CO to C3fuel using asymmetric C–C coupling active sites., 2019, 10(1): 5186.
[26] MA M, DJANASHVILI K, SMITH W A. Selective electrochemical reduction of CO2to CO on CuO-derived Cu nanowires., 2015, 17(32): 20861–20867.
[27] LIU G, LI Z, SHI J,. Black reduced porous SnO2nanosheets for CO2electroreduction with high formate selectivity and low overpotential., 2019, 260: 118134.
[28] CHOU T C, CHANG C C, YU H L,. Controlling the oxidation state of Cu electrode and reaction intermediates for electrochemical CO2reduction to ethylene., 2020, 142: 2857–2867.
[29] DAIYAN R, SAPUTERA W H, ZHANG Q,. 3D heterostructured copper electrode for conversion of carbon dioxide to alcohols at low overpotentials., 2019, 3(1): 1800064.
Modulation of CuO Surface Properties for Selective Electrocatalytic Reduction of CO2to HCOOH
GUO Lina, HE Xuebing, LYU Lin, WU Dan, YUAN Hong
(Key Laboratory of Pesticide and Chemical Biology, Ministry of Education, College of Chemistry, Central China Normal University, Wuhan 430079, China)
The electrocatalytic carbon dioxide reduction reaction can convert the greenhouse gas carbon dioxide into chemical raw materials or organic fuels, providing a feasible way to overcome global warming and the conversion of electrical energy to chemical energy. The main challenge of this technology is the wide product distribution, resulting in low selectivity of a single product, however, modulating the surface properties of the catalyst is an efficient strategy to solve this problem. In this study, the precursors of Cu2O and Cu2S were oxidized to the CuO catalysts with different surface properties. The CuO-FS catalyst derived from Cu2S delivered the improved activity of electro-reduction of carbon dioxide and selectivity for formic acid product. This catalyst exhibited a higher total current density and the Faraday efficiency of formic acid > 70% in a wide test voltage range of –0.8 – –1.1 V; the Faraday efficiency for formic acid could reach a maximum of 78.4% at –0.9 V. The mechanism study indicated that the excellent performance of CuO-FS for electro-reduction of carbon dioxide could be attributed to the large electrochemically active surface area, which provided a large number of surface active sites, resulting in a higher total current density; moreover, the less zero-valent Cu was produced over the surface of CuO-FS during the electrocatalytic process, which reduced the production of ethylene and thus promoted the production of formic acid.
CO2electroreduction; Cu-based catalyst; surface properties; product selectivity; formic acid
1000-324X(2022)01-0029-09
10.15541/jim20210547
TQ151
A
2021-09-04;
2021-10-22;
2021-11-01
软物质材料化学与功能制造重庆市重点实验室开放经费(20191002)
Chongqing Key Laboratory of Soft-Matter Material Chemistry and Function Manufacturing (20191002)
郭李娜(1998–), 女, 硕士研究生. E-mail: gln@mails.ccnu.edu.cn
GUO Lina (1998–), female, Master candidate. E-mail: gln@mails.ccnu.edu.cn
原弘, 教授. E-mail: yuanhong@mail.ccnu.edu.cn
YUAN Hong, professor. E-mail: yuanhong@mail.ccnu.edu.cn
猜你喜欢
油气田地面工程(2022年8期)2022-10-02
分析化学(2018年12期)2018-01-22
有色金属材料与工程(2017年4期)2017-09-18
作文周刊·七年级版(2016年47期)2017-05-31
科技创新与应用(2017年11期)2017-04-27
分析化学(2017年1期)2017-02-06
中小企业管理与科技·下旬刊(2016年12期)2017-01-17
决策(2016年1期)2016-09-10
科技资讯(2015年8期)2015-07-02
故事会(2015年12期)2015-05-14
