
改性氮化碳光催化剂在生物质氧化反应中的应用
2022-04-12 10:43刘雪晨曾滴周沅逸王海鹏张玲王文中
无机材料学报 2022年1期
刘雪晨, 曾滴,2, 周沅逸,2, 王海鹏,2, 张玲,2, 王文中,2,3
改性氮化碳光催化剂在生物质氧化反应中的应用
刘雪晨1, 曾滴1,2, 周沅逸1,2, 王海鹏1,2, 张玲1,2, 王文中1,2,3
(1. 中国科学院 上海硅酸盐研究所, 上海 200050; 2. 中国科学院大学, 北京 100049; 3. 中国科学院大学 杭州高等研究院, 杭州 310024)
利用氮化碳光催化剂催化生物质选择性转化, 不仅扩展了非金属催化剂的应用领域, 而且能够缓解化工产品过度依赖于化石能源的现状。2,5-二甲酰基呋喃是生产多种化工产品的关键中间体, 本研究将均苯四甲酸二酐引入氮化碳骨架, 并利用H2O2进行处理, 制备了含有氮羟基的改性氮化碳光催化剂, 并探究其在可见光激发下将生物质平台分子5-羟甲基糠醛通过绿色化学的方法选择性地氧化为2,5-二甲酰基呋喃的性能。结果表明:经过H2O2改性的催化剂, 在可见光激发下可以产生氮氧自由基, 使得底物分子侧链上的羟基选择性地氧化为醛基, 避免了在水相光催化条件下可能产生的多种活性氧物种引起的开环、矿化反应等副反应。特别是, 当光催化剂前驱体中蜜勒胺与均苯四甲酸二酐的比例为1 : 2时, 在400 nm LED光源激发下, 目标产物的选择性可达到96.2%。
光催化; 5-羟甲基糠醛; 2,5-二甲酰基呋喃; 氮氧自由基; 氮化碳
在众多光催化剂中, 氮化碳材料作为一种非金属半导体催化剂, 具有成本低廉、环境友好和易于回收等优点[1], 在光催化领域已得到广泛应用[2], 如光催化固氮[3]与光催化染料降解[4]等。近年来光催化剂在生物质的选择性转化领域的应用逐渐成为研究热点, 其反应条件温和, 无需高压氧气条件, 反应后排放物对环境友好, 具有良好的发展前景[5]。将来源于自然界的可再生的生物质转化为化工原料, 能够替代传统的石油衍生物[6]。将生物质平台分子5-羟甲基糠醛(HMF)选择性地转化为2,5-二甲酰基呋喃(DFF)是生物质升值转化过程中的重要反应之一, 后者是生产药物、杀菌剂及生物基聚合物不可或缺的中间体[7]。
HMF分子含有一个呋喃环, 并且两边侧链各有一个羟基和一个醛基, 化学性质非常活泼[8]。已有研究使用氮化碳光催化剂氧化呋喃类醇多在有机介质中进行, Krivtsov等[9]首次利用热剥离法得到TE-g-C3N4光催化剂, 并在水相体系中氧化5-羟甲基糠醛至2,5-二甲酰基呋喃, 但选择性仅为45%。在绿色水相体系中, 催化剂易产生强氧化性物种, 导致呋喃环发生开环反应[10], 甚至深度矿化底物为CO2和H2O[8], 限制了目标产物的选择性。因此, 寻找合适的光催化材料高选择性地转化合成DFF依然是生物质转化中的一个重要挑战。
本研究将均苯四甲酸二酐(PMDA)引入氮化碳材料的结构中[11-12], 使用双氧水对其进行改性, 通过提高光催化材料价带电位, 提高其空穴氧化能力, 另外通过在催化剂表面引入活性的氮羟基基团, 使其在光引发下可以转化为对醇分子具有选择性氧化能力的氮氧自由基。基于此, 本研究在可见光激发下, 以空气中的氧为氧化反应助剂, 在水溶液反应体系中将HMF高选择性地转化为目标产物, 结合对催化剂的各种表征以及反应中各种氧化物种对选择性氧化反应影响的分析, 提出一种可能的氮氧自由基参与的催化氧化机制, 为氮化碳材料的设计与改性提供新的思路。
1 实验方法
1.1 样品制备
以三聚氰胺与均苯四甲酸二酐(PMDA)合成改性氮化碳材料(PI)。首先, 将10 g三聚氰胺置于坩埚中, 使用马弗炉在425 ℃保温4 h制备得到蜜勒胺(melem)。制备得到的粉末样品与均苯四甲酸二酐在玛瑙研钵中等摩尔比混合后在325 ℃马弗炉中保温4 h, 取出, 使用去离子水清洗后60 ℃干燥, 得到PI(1:1)。改变两种前体的混合比例(1:2与2:1)所制备得到的样品分别命名为PI(1:2)与PI(2:1)。
取1 g制备得到的PI(1:2)材料加入三颈烧瓶中, 再加入20 mL的H2O2溶液(浓度≥30%), 置于油浴锅中升温至120 ℃后继续保温1 h, 使用去离子水清洗后60 ℃干燥, 得到PI(1:2)-120。改变PI材料用量, 制备步骤同上得到PI(1:1)-120。将10 g三聚氰胺置于坩埚中, 在550 ℃马弗炉中保温4 h, 取出, 去离子水清洗后60 ℃干燥, 得到C3N4。
1.2 样品表征
通过Rigaku MiniflexⅡ台式X射线衍射仪表征样品的结晶性能, X 射线为Cu靶的Kα射线, 测试范围是10°≤2≤60°; 利用Lambda FTIR-7600光谱仪在4000~400 cm–1范围内采集样品的红外光谱; 利用Hitachi U-3010分光光度计测定样品的UV-Vis 漫反射光谱(DRS); 在JEM-F2500透射电子显微镜(TEM)上采集样品图像; 在ThermoESCALAB 250Xi型X射线光电子能谱仪(XPS)上测试样品的XPS图谱; 在Auto Chem Ⅱ 2920仪器上测量氧气程序升温脱附(O2-TPD); 室温下, 以320 nm为激发波长, 在 Hitachi F-4600荧光分光光度计上测量样品的光致发光光谱(PL); 在Thermo Scientific Nicolet iS10红外光谱仪上原位采集样品的傅立叶变换红外光谱, 灯源选择300 W氙灯; 在上海辰华公司的CHI660D电化学工作站进行样品的电化学测试,采用三电极体系在0.1 mol/L的Na2SO4溶液(pH=6.8)中进行电化学测试, 使用500 W Xe灯(CHF-XM500)作为光源。
1.3 光催化氧化性能研究
在具有石英窗口玻璃反应器中测试样品的光催化性能, 反应使用300 W氙灯(均使用滤波片滤去<300 nm的光)与400 nm LED灯。反应操作过程如下:在30 mL不同浓度的HMF水溶液中, 均匀分散50 mg催化剂。在暗态条件下, 持续搅拌30 min, 使HMF与催化剂之间达到吸附-解吸平衡后, 开灯光照开始氧化反应。在适当的时间间隔取出1 mL溶液过滤去除粉末催化剂, 通过高效液相色谱(Agilent C18色谱柱)分析溶液中各物质的浓度。
2 结果与讨论
2.1 光催化剂的表征
图1为不同制备条件下获得样品的XRD谱图。图1(a)为PI样品、C3N4样品与蜜勒胺、均苯四甲酸二酐前体的XRD图谱。PI(1:1)在2=15°~30°范围内出现了与前体均不重合的新峰, 说明PI样品中具有三种结构单元:2=19.0°处的衍射峰对应于均苯四甲酸二酰亚胺(PDI)与其自身形成的结构单元, 即PDI-PDI; 2=27.4°处的衍射峰对应于蜜勒胺与其自身形成的结构单元, 即melem-melem; 而2=29.6°处的衍射峰则对应PDI-melem复合的结构单元[13]。图1(b)显示, 当其他制备条件不变时, 改变前体的摩尔比例, PI样品的结晶度会发生显著的变化。当蜜勒胺与均苯四甲酸二酐的摩尔比例为1 : 2时, PI(1:2)样品具有最明显的衍射峰, 说明其结晶度最高; 当蜜勒胺与均苯四甲酸二酐的摩尔比例为1 : 2与1:1时, 从样品的XRD图谱中能观察到三种不同结构单元的衍射峰, 但PI(1:1)的PDI-melem复合结构单元衍射峰减小。当均苯四甲酸二酐的摩尔比例进一步降低时, 样品在2=29.6°处的衍射峰几乎完全消失, 同时在2=10°~15°范围内出现蜜勒胺的衍射峰, 说明此条件下得到的样品中不存在PDI-melem的复合结构, 且存在部分没有参与结合的蜜勒胺单体[14]。此外, 实验对比了经过120 ℃ H2O2处理后PI(1:2)的结构变化:PDI-melem的衍射峰的整体强度有所下降, 而2=19.0°, 27.4°处的衍射峰强度增强, 说明melem-melem之间和PDI-PDI之间结构相对稳定, H2O2处理主要导致PDI-melem之间结构的断裂。
利用TEM表征PI(1:2)及PI(1:2)-120样品的形貌变化。从图2可以看出, 两种样品均为片状结构, 经H2O2处理后, 样品的孔隙数量增加。图2(d)更清晰地显示出PI(1:2)-120疏松多孔的形貌特征。结合XRD谱图的分析结果, 可以推测合成的PI样品经H2O2进行处理后, 部分结构被分解, 断裂处形成大量孔洞, 造成了催化剂形貌结构的改变。
利用X射线光电子能谱(XPS)进一步表征了制备样品的化学成分。如图3(a~d)所示, 以PI(1:1)为例, 285.1 eV的C1s峰来源于均苯四甲酸二酰亚胺结构中苯环(C=C)上的碳原子, 而288.0和288.6 eV的C1s峰分别对应三嗪环(C=N)中的碳原子和亚胺的羰基(C=O)碳原子[15]。与PI(1:1)对比, PI(1:2) 285.1 eV处的峰明显增强, 说明PI(1:2)中具有更高比例的PDI结构。经过H2O2处理后, PI样品的苯环C1s峰均明显减小, 且均苯四甲酸二酰亚胺中的羰基C1s峰均向高结合能偏移, 说明H2O2处理使部分PDI结构脱离与PI有序结构的连接。
O2-TPD的谱图可以反映催化剂对O2的吸附能力[16]。如图4(a), PI(1:2)仅在115 ℃下出现一个氧解吸峰, 而PI(1:2)-120在102和250 ℃下表现出两个氧解吸峰, 说明经过H2O2处理后, 催化剂的O2吸附能力增强。图4(b)为不同样品的紫外-可见漫反射(UV-Vis)光谱, 与C3N4相比, 不同前体比例的PI样品在450~800 nm可见光波段的吸收均有提高, 经过H2O2处理后的PI(1:2)催化剂在400~500 nm波段吸收进一步增强。
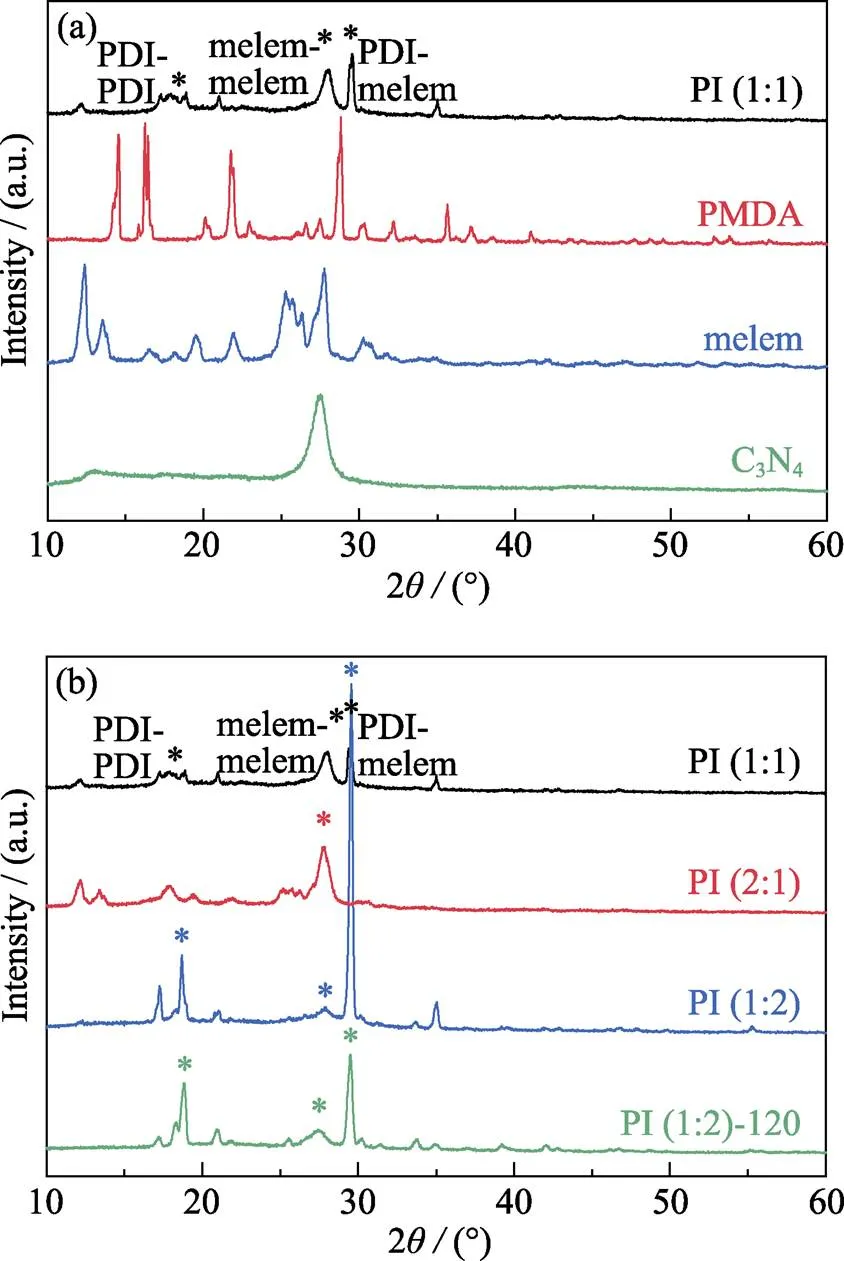
图1 不同制备条件下获得样品的XRD谱图
(a) Sample PI (1:1) and its precursor PMDA and melem; (b) Samples PI (1:1), PI (2:1), PI (1:2), and PI (1:2)-120

图2 样品PI(1:2)(a, b)和样品PI(1:2)-120(c, d)的TEM照片
图4(c)为光照下催化剂在FTO电极上的光电流响应, PI(1:2)-120与PI(1:2)的光电流响应都显著高于C3N4。但与PI(1:2)相比, 处理后的PI样品光电流响应较弱, 这可能是由于PI(1:2)-120在500~ 800 nm可见光区域的光吸收较弱[17]。除了光电流响应的强弱, 光生载流子的复合时间也是反映光催化剂光电性能的一个重要因素, 图S1给出了瞬时光电流的衰减率, 对比了不同催化剂的电荷重组行为[18]。PI(1:2)-120的瞬时光电流的衰减比PI(1:2)明显更缓慢, 说明其电荷重组较慢, 光催化活性更高。图S2中根据Mott-Schottky图, 催化剂的光吸收特性以及VB XPS的表征, 得出了C3N4、PI(1:2)以及PI(1:2)-120催化剂的能带位置示意图。
2.2 光催化性能测试及影响因素分析
不同催化剂催化HMF氧化反应的性能汇总于表1, 第3~5行显示随着前驱体中均苯四甲酸二酐比例的增加, 氧化反应的转化率下降, 而选择性逐渐增加, 不同前体比例催化剂的性能变化可能与其结晶度的差异有关。用H2O2对样品进一步处理后, 反应目标产物的选择性显著提升, 以PI(1:2)为例, 选择性由19.3%增至67.8%。在前期的实验探究中发现, 小于400 nm波长的光照易将HMF转化为开环副产物。选择400 nm LED灯与性能最优的PI(1 : 2)-120样品进一步进行实验, 反应12 h后, 转化率超过50%, 且其选择性能够达到96.2%。

图3 不同样品的XPS谱图(C1s)(a~d)和PI样品的结构简图(e)(不同颜色的C原子与XPS谱图对应)
(a) PI (1:1); (b) PI (1:1)-120; (c) PI (1:2); (d) PI (1:2)-120; Colorful figures are available on website

图4 不同样品的O2-TPD图(a), 紫外-可见漫反射图(b)及光电流图谱(c)
Colorful figures are available on website

表1 不同催化剂对HMF催化氧化的实验结果
[a] Experimental conditions: 50 mg catalyst, 300 W Xe lamp, 30 mL deionized water, 3.33 mmol/L HMF, atmospheric pressure air atmosphere, 15 ℃, reaction time: 3 h;
[b] Experimental conditions: 400 nm LED lamp, 30 mL 0.67 mmol/L HMF aqueous solution, 15 ℃, reaction time: 12 h;
[c] Experimental conditions were the same as [a], with 100 μL of 30% H2O2added before the reaction; Con: conversion; Sel: selectivity
实验检测了不同PI样品在水溶液中所产生的活性氧物种。超氧自由基(O2–)能够将氮蓝四唑(NBT)还原为蓝色甲臜, 因此通过检测反应后NBT水溶液中甲臜的含量, 能够对比催化剂产生超氧自由基的能力。由图S3(a)所示, PI(1:2)的吸光度高于PI(1:2)-120样品, 说明在光照反应过程中PI(1:2)产生的超氧自由基更多。如图S3(b)所示, 在对苯二甲酸水溶液中对比了两种催化剂光照下羟基自由基产量, 与PI(1:2)相比, PI(1:2)-120样品产生的羟基自由基更少。进一步定量对比两种催化剂中H2O2的产量, 如图S3(c), 光照反应1 h, 两者分别产生8.6、32.8 μmol的H2O2。此外, 如果PI(1:2)不经过前期的H2O2处理, 而是直接在反应体系中添加双氧水反而会显著降低DFF的选择性(表1, 第9行), 由此说明H2O2并不是作为氧化剂提升反应的性能, 而是通过在一定温度下对催化剂本身的改性来提升催化体系中目标产物的选择性。结合催化剂对HMF实验结果与氧化物种的测试, PI(1:2)-120样品在光照下产生的非选择性氧化物种减少对应于目标产物DFF选择性的显著提升。
半导体催化剂中存在激子效应, 三线态激子与基态氧分子之间的共振能量转移能够促进单线态氧产生, 使其进一步参与催化氧化反应[19-21]。因此, 本研究同样考虑了在改性氮化碳催化剂中激子效应对于催化性能的潜在作用。根据已有文献[1]的报道, 单线态氧将选择性氧化HMF分子至2,5-呋喃二甲酸, 而非2,5-二甲酰基呋喃[1]。在前述实验条件下, 使用H2O2处理前后的改性氮化碳为催化剂, 但产物中均未检测到2,5-呋喃二甲酸, 反映此催化模型中没有明显的激子效应对于催化性能的影响。
2.3 氮氧自由基的检测及催化机理分析
综合材料表征与性能测试, 推测PI催化剂经过H2O2处理后, 其有序结构被破坏, 主要发生了两种结构单元之间连接的断裂。断裂后, 暴露的酰亚胺基团中的仲胺被H2O2高温处理转化为相应的氮羟基化合物。据文献[22-25]报道, 氮羟基化合物在超氧自由基或空穴的活化下能够生成具有选择性氧化能力的氮氧自由基。因此, 对催化剂PI(1:2)-120表面是否存在氮羟基, 以及在光激发下是否可能产生氮氧自由基进行了表征。
如图5(a), 经H2O2处理的PI(1:2)-120样品的红外光谱中, C=O双键对应的724、1712与1774 cm–1峰强度均有所增加[26], 并且1360 cm–1处信号增强, 对应于催化剂表面N–OH的振动[27]。结合催化剂的表征结果, 经过H2O2处理后, 催化剂的melem-PDI结构减少, 即断裂主要发生在米勒胺与四甲酸二酐的连接部位, 而四甲酸二酐中的羰基没有受到H2O2的影响。原位红外光谱通过持续通入湿润空气, 模拟催化反应进行时的反应条件, 并间隔一定的光照时间点, 进一步表征了PI催化剂在光照射下的变化。如图5(b), 3000~3500 cm–1处的宽峰来源于吸附水, 由于通入的是湿润空气, 水的信号峰不可避免。但能够观察到, 随着光照时间的延长, 3000~ 3500 cm–1处峰的宽度逐渐增加, 并且峰形发生改变, 由单一宽峰逐渐分裂为两个峰。根据文献[28], 未活化的N–OH与活化后的N–O·之间存在氢键的相互作用, 红外谱图中能够观察到3250 cm–1处信号增强。此外, 使用紫外-可见分光光度计测量特定波长处的吸收变化能够表征溶液中氮氧自由基的浓度变化[29]。根据已有文献[30]报道, 水溶液中氮氧自由基形成后在=385 nm处产生一个吸收峰, 吸收峰值的大小与氮氧自由基的浓度呈正相关。本研究将4 mmol/L的HMF水溶液滴加到分散有催化剂的比色皿中, 检测了=385 nm处吸光度随时间的变化情况。图5(c)显示出催化剂在水溶液中产生的N–O·浓度随时间的变化曲线, 未经过处理的PI(1:2)催化剂对应的曲线没有明显的变化, 说明其中没有产生N–O·; 经过H2O2改性后的PI(1:2)-120催化剂水溶液中N–O·浓度逐渐上升并最终达到平衡。

图5 氮氧自由基的检测及催化机理分析
(a) FT-IR spectra of PI (1:2) and PI (1:2)-120; (b)FT-IR spectra of PI (1:2)-120 illuminated in air; (c) Absorption intensitytime for catalyst suspensions with 4 mmol/L HMF added

图6 PI催化剂在H2O2处理前后可能的结构变化及其催化氧化HMF可能的机制
结合催化剂的结构(表面具有氮羟基)及在光催化条件下能产生光生载流子的特点, 本研究提出PI(1:2)-120催化氧化HMF的可能机制(如图6):在光照下, 催化剂产生光生电子与空穴, 其中空穴转移至氮羟基化合物结构中, 活化N–OH为N–O自由基, 同时放出的质子与O2和光生电子共同作用转化为H2O2[31]。氮氧自由基能够选择性地氧化羟基[29], 可以将反应底物的羟甲基基团氧化至醛基, 并在氧化过程中夺取质子, 重新回到N–OH状态[25]。PI(1:2)-120在光照下利用氮氧自由基实现选择性氧化, 避免了过氧化导致的反应底物分解, 这是其选择性高的重要原因。
通过重复实验对催化剂的循环性能进行了表征(图S4), 从图中可以看出, 在前四轮循环实验中, 反应底物的转化率基本维持在同一水平(53.3%~ 59.9%), 产物的选择性则呈现下降趋势, 由96.2%降至54.6%。通过催化剂再生能够很好地解决选择性下降的问题, 在120 ℃下对用H2O2将经过四轮反应后的催化剂重新处理, 经过清洗烘干后用于第五轮循环测试。结果显示, 经过H2O2再处理后, 失活催化剂的催化活性能够恢复到至接近初始水平, 反应达到82.9%的高选择性。
3 结论
将5-羟甲基糠醛高选择性地催化氧化生成2,5-二甲酰基呋喃, 对于基于生物质的绿色化工发展具有重要的实际意义。通过改变催化剂组分及H2O2处理, 获得了高性能改性氮化碳催化剂PI(1:2)-120, 在400 nm的LED光照下, 能够实现高于96%的DFF选择性转化。结合对催化剂结构的表征、催化剂的催化性能、活性物种的测试结果及原位表征, 提出了一种可能的反应机理, 即处理后催化剂在光激发下能够产生氮氧自由基参与选择性氧化过程。通过原位引入生成的氮氧自由基在绿色水相体系中实现了生物质平台分子的高选择性转化, 并拓展了氮化碳材料的改性方法。
与本文相关的补充材料可登录https://doi.org/ 10.15541/jim20210262查阅。
[1] XU S, ZHOU P, ZHANG Z,Selective oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid using O2and a photocatalyst of co-thioporphyrazine bonded to g-C3N4., 2017, 139(41): 14775–14782.
[2] NENG L, ZHOUZHOU K, XINGZHU C,Research progress of novel two-dimensional materials in photocatalysis and electrocatalysis., 2020, 35(7): 735– 747.
[3] XIAO C, ZHANG L, WANG KA new approach to enhance photocatalytic nitrogen fixation performancephosphate-bridge: a case study of SiW12/K-C3N4., 2018, 239: 260–267.
[4] ZHANG S, ZOU Y T, CHEN Z S,Visible-light-driven activation of persulfate by RGO/g-C3N4composites for degradation of BPA in wastewater., 2020, 35(3): 329–336.
[5] WU Q, HE Y, ZHANG H,Photocatalytic selective oxidation of biomass-derived 5-hydroxymethylfurfural to 2,5-diformylfuran on metal-free g-C3N4under visible light irradiation., 2017, 436: 10–18.
[6] CHENG L, ZHANG Y, ZHU Y,Selective oxidation of HMF., 2021, 33(2): 318–330.
[7] HU L, LIN L, WU Z,Recent advances in catalytic transformation of biomass-derived 5-hydroxymethylfurfural into the innovative fuels and chemicals., 2017, 74: 230–257.
[8] YURDAKAL S, TEK B S, ALAGÖZ O,Photocatalytic selective oxidation of 5-(hydroxymethyl)-2-furaldehyde to 2,5-furandicarbaldehyde in water by using anatase, rutile, and brookite TiO2nanoparticles., 2013, 1(5): 456–461.
[9] KRIVTSOV I, GARCíA-LÓPEZ E I, MARCì G,Selective photocatalytic oxidation of 5-hydroxymethyl-2-furfural to 2,5-furandicarboxyaldehyde in aqueous suspension of g-C3N4., 2017, 204: 430–439.
[10] LI S, SU K, LI Z,Selective oxidation of 5-hydroxymethylfurfural with H2O2catalyzed by a molybdenum complex., 2016, 18(7): 2122–2128.
[11] CHU S, PAN Y, WANG Y,Polyimide-based photocatalysts: rational design for energy and environmental applications., 2020, 8(29): 14441–14462.
[12] CHU S, WANG Y, GUO Y,Band structure engineering of carbon nitride: in search of a polymer photocatalyst with high photooxidation property., 2013, 3(5): 912–919.
[13] KOFUJI Y, ISOBE Y, SHIRAISHI Y,Carbon nitride- aromatic diimide-graphene nanohybrids: metal-free photocatalysts for solar-to-hydrogen peroxide energy conversion with 0.2% efficiency., 2016, 138(31): 10019–10025.
[14] YANG C, FOLENS K, DU LAING G,Improved quantum yield and excellent luminescence stability of europium-incorporated polymeric hydrogen-bonded heptazine frameworks due to an efficient hydrogen-bonding effect., 2020, 30(39): 2003656.
[15] CUI Z, ZHOU J, LIU T,Porphyrin-containing polyimide with enhanced light absorption and photocatalysis activity., 2019, 14(12): 2138-2148.
[16] BAI J, SUN Y, LI M,The effect of phosphate modification on the photocatalytic H2O2production ability of g-C3N4catalyst preparedacid-hydrothermal post-treatment., 2018, 87: 1–9.
[17] KOFUJI Y, OHKITA S, SHIRAISHI Y,Graphitic carbon nitride doped with biphenyl diimide: efficient photocatalyst for hydrogen peroxide production from water and molecular oxygen by sunlight., 2016, 6(10): 7021–7029.
[18] NG Y H, IWASE A, KUDO A,Reducing graphene oxide on a visible-light BiVO4photocatalyst for an enhanced photoelectrochemical water splitting., 2010, 1(17): 2607–2612.
[19] WANG H, JIANG S, CHEN S,Insights into the excitonic processes in polymeric photocatalysts., 2017, 8(5): 4087–4092.
[20] HUI W, DONG Z X, YI X. Recent progresses on the photoexcitation processes of polymeric carbon nitride-based materials., 2017, 33(11): 1897–1913.
[21] WANG H, JIANG S, CHEN S,Enhanced singlet oxygen generation in oxidized graphitic carbon nitride for organic synthesis., 2016, 28(32): 6940–6945.
[22] ZHANG C, HUANG Z, LU J,Generation and confinement of long-lived-oxyl radical and its photocatalysis., 2018, 140(6): 2032–2035.
[23] ZHAO G, HU B, BUSSER G W,Photocatalytic oxidation of alpha-C-H bonds in unsaturated hydrocarbons through a radical pathway induced by a molecular cocatalyst., 2019, 12(12): 2795–2801.
[24] KASHPAROVA V P, KLUSHIN V A, LEONTYEVA D V,Selective synthesis of 2,5-diformylfuran by sustainable 4-acetamido- TEMPO/halogen-mediated electro-oxidation of 5-hydroxymethylfurfural., 2016, 11(18): 2578–2585.
[25] LIU G, TANG R, WANG Z. Metal-free allylic oxidation with molecular oxygen catalyzed by g-C3N4and-hydroxyphthalimide., 2014, 144(4): 717–722.
[26] KOFUJI Y, OHKITA S, SHIRAISHI Y,Mellitic triimide- doped carbon nitride as sunlight-driven photocatalysts for hydrogen peroxide production., 2017, 5(8): 6478–6485.
[27] HU K, TANG J, CAO S,TEMPO-Functionalized aromatic polymer as a highly active, pH-responsive polymeric interfacial catalyst for alcohol oxidation., 2019, 123(14): 9066–9073.
[28] FRANCHI P, LUCARINI M, PEDRIELLI P,Nitroxide radicals as hydrogen bonding acceptors. an infrared and EPR study., 2002, 3: 789–793.
[29] KOMPANETS M O, KUSHCH O V, LITVINOV Y E,Oxidation of 5-hydroxymethylfurfural to 2,5-diformylfuran with molecular oxygen in the presence of-hydroxyphthalimide., 2014, 57: 60–63.
[30] RECUPERO F, PUNTA C. Free radical functionalization of organic compounds catalyzed by N-hydroxyphthalimide., 2007, 107: 3800–3842.
[31] HAO H, SHI J L, XU H,-hydroxyphthalimide-TiO2complex visible light photocatalysis., 2019, 246: 149–155.
Selective Oxidation of Biomass over Modified Carbon Nitride Photocatalysts
LIU Xuechen1, ZENG Di1,2, ZHOU Yuanyi1,2, WANG Haipeng1,2, ZHANG Ling1,2, WANG Wenzhong1,2,3
(1. Shanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai 200050, China; 2. University of Chinese Academy of Sciences, Beijing 100049, China; 3. Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, Hangzhou 310024, China)
Using carbon nitride photocatalysts to catalyze the selective conversion of biomass platform molecules could not only expand the application fields of non-metallic catalysts but also alleviate the dependence on fossil energy in chemical manufacturing. 2,5-diformylfuran is an indispensable intermediate for the production of value-added chemicals. In this study, pyromellitic dianhydride and melem were used as the precursors. The pyromellitic dianhydride monomer was incorporated into the carbon nitride skeleton by high-temperature heat-treatment, and the catalysts were further treated with H2O2. Finally non-metallic carbon nitride photocatalysts containing nitrogen hydroxyl groups were prepared. Moreover, its performance in the selective oxidation of 5-hydroxymethylfurfural into 2,5-diformylfuran under visible light excitation was investigated. The results showed that H2O2-treated samples could generate nitroxide radicals under light irradiation, resulting in the selective oxidation of hydroxyl groups on the side chains of 5-hydroxymethylfurfural molecules into aldehyde groups, avoiding undesired side-reactions (ring-opening, mineralization reactions,.) caused by various reactive oxygen species that may be generated in aqueous solutions. In particular, under the excitation of an LED light source with a specific wavelength (400 nm), when the ratio of melem to PMDA precursor was 1 : 2, the selectivity of 2,5-diformylfuran over the photocatalyst could reach 96.2%.
photocatalysis; 5-hydroxymethylfurfural; 2,5-diformylfuran; nitroxide radical; carbon nitrde
1000-324X(2022)01-0038-07
10.15541/jim20210262
O643
A
2021-04-20;
2021-06-23;
2021-06-30
国家自然科学基金(51972327, 51972325)
National Natural Science Foundation of China (51972327, 51972325)
刘雪晨(1996–), 女, 硕士研究生. E-mail: xuechen1215@shu.edu.cn
LIU Xuechen(1996–), female, Master candidate. E-mail: xuechen1215@shu.edu.cn
张玲, 副教授. E-mail: lingzhang@mail.sic.ac.cn; 王文中, 教授. E-mail: wzwang@mail.sic.ac.cn
ZHANG Ling, associate professor. E-mail: lingzhang@mail.sic.ac.cn;
WANG Wenzhong, professor. E-mail: wzwang@mail.sic. ac.cn
猜你喜欢
疯狂英语·新阅版(2021年9期)2021-10-30
科学与财富(2021年13期)2021-07-04
电脑知识与技术·经验技巧(2020年7期)2020-08-23
中国科技纵横(2019年3期)2019-03-25
分析化学(2017年12期)2017-12-25
有色金属材料与工程(2016年6期)2017-05-31
科教导刊·电子版(2016年25期)2016-11-16
分析化学(2015年3期)2015-04-20
湖北农业科学(2014年12期)2015-01-06
中小企业管理与科技·中旬刊(2014年7期)2014-09-24
