
特发性肺纤维化相关的微阵列鉴定基因和microRNA分析
2022-03-30 04:52姜淑娟杨栋梁
河北医科大学学报 2022年3期
宋 鹏,张 猛,姜淑娟,杨栋梁
(1.山东第一医科大学附属省立医院呼吸与危重症医学科,山东 济南 250000;2.山东省济南市人民医院全科医学科, 山东 济南 271100;3.沧州医学高等专科学校公共课教学部,河北 沧州 061000)
特发性肺纤维化(idiopathic pulmonary fibrosis,IPF)是特发性间质性肺炎最常见的形式,其特征在于临床症状为咳嗽和呼吸困难,气体交换受限以及进行性肺瘢痕形成[1]。目前己批准2种用于治疗IPF的有效药物(尼达尼布和吡非尼酮)[2-3]。但是,由于药物的可及性及疗效的限制,大多数IPF患者最终死于呼吸衰竭。IPF的预后仍然充满挑战,因此需要更加全面地了解其疾病发病机制。微小RNA(microRNA,miRNA)是一类非编码RNA,可以通过与mRNA结合来抑制靶基因的表达[4]。 由于其在细胞凋亡、增殖和分化等细胞过程中的关键作用而引起了广泛关注。在过去的十年中越来越多的研究寻找IPF潜在的生物标志物,多种miRNA可以作为IPF预测的潜在生物标记,包括miR-92a、miR-210、miR-29、miR-326、miR-98 和miR-let-7d[5-7]。但是单个miRNA的研究不能满足IPF快速发展的要求,构建miRNA-mRNA联合网络可能有助于探索适用于临床的生物标记物和治疗靶标的进一步开发。本研究旨在通过基因表达综合(gene expression omnibus,GEO)数据库鉴定miRNA表达谱中的差异表达miRNA(differentially expressed miRNAs,DEMis)和差异表达mRNA(differentially expressed mRNAs,DEMs),以探索IPF的生物学过程。使用全面的生物信息学分析检查DEMs和DEMis之间的相关性,并结合从DEMis和DEMs数据中检索到的信息,通过GO及KEGG途径富集分析筛选出IPF的潜在生物标志物。
1 资 料 与 方 法
1.1数据集的获取和分析 IPF相关miRNA和mRNA表达谱中的微阵列数据可从GEO数据库(http://www.ncbi.nlmNih.gov/geo)中检索和下载。使用“idiopathic pulmonary fibrosis”作为关键字进行查询。搜索仅限于以下特定领域:研究类型,按阵列进行的表达谱分析和种类(智人)。下载miRNA表达微阵列数据集GSE27430和mRNA表达微阵列数据集GSE135065。使用R studio中的limma软件包获得IPF患者与健康对照组之间差异表达的miRNA和mRNA。贝叶斯方法纠正批量效应。如果有多个探针映射到同一基因中,则平均表达值被用来等于该基因的表达值。采用t检验过滤差异表达的基因。DEMis和DEMs通过P<0.05和log FC>平均(logFC)+ 2×SD(logFC)进行筛选。为了显示DEMis和DEMs在不同样品中的差异表达,通过在R studio中应用图和Pheatmap包绘制火山图和热图。
1.2转录因子 靶标的GO富集分析将DEMis上传到FunRich(3.1.3),这是用于对转录因子途径的富集靶标(基因/mRNA)进行GO功能富集分析的常用工具。GO富集分析还用于开发miRNA、基因/mRNA和转录因子之间的相互作用网络分析。
1.3DEMis靶向mRNA的预测 使用miRDB(http://www.mirdb.org)、miRTarBase(http://mirtarbase.mbc.nctu.edu.tw)和TargetScan(http://www.targetscan.org)数据库来预测DEMis的靶向mRNA。之后,通过匹配之前选择的DEMs进一步过滤DEMis的预测mRNA,然后得到DEMis-DEMs的配对组合。
1.4miRNA-mRNA调控网络的构建 将上面选择的所有DEMis-DEMs通过配对来构建miRNA-mRNA网络,同时使用Cytoscape软件(3.7.2版)对其进行可视化。
1.5网络中mRNA的GO和KEGG富集分析 在R Studio中使用了richplot和ggplot2软件包对网络中的mRNA进行GO和KEGG途径分析。
1.6统计学方法 应用R语言V3.6.0 统计软件分析数据。计量资料比较采用t检验,为了进一步控制错误率,使用了Benjamini和Hochberg方法来计算调整后的P值。P<0.05为差异有统计学意义。
2 结 果
2.1IPF中的DEMis筛选结果 根据前所述排除标准,miRNA的log FC的临界值为1.5。确定了21个上调或下调的miRNAs,包括14个上调的miRNA以及7个下调的miRNA,见表1。通过log10(P值)和log(FC)相关性的火山图直观地说明了IPF和健康对照之间miRNA表达差异的分布(图1)。并绘制热图以直观显示IPF和健康对照组之间的差异(图2)。

图1 DEMis的火山图。红点:上调的基因;绿点:下调的基因

图2 DEMis的热图。左侧12个样品来自对照组,右侧13个样品来自IPF组;红色:高表达; 蓝色:低表达
2.2差异miRNA的转录因子富集分析 32个转录因子中总共映射748个基因。通过转录因子靶标的富集,筛选了与miRNA密切相关的前10个转录因子,分别为特异性蛋白1(specific protein 1,SP1)、早期生长应答因子1(early growth response 1,EGR1)、NK6同源框蛋白1重组蛋白(recombinant NK6 homeobox protein 1,NKX6-1)、结构域 2 类转录因子 1(poudomain class 2 transcriptionfactor 1,POU2F1)、富含 AT 的交互式含域蛋白 3A(AT-rich interactive domain-containing protein 3,ARID3A)、心肌细胞特异性增强因子2A重组蛋白(recombinant myocyte specific enhancer factor 2A,MEF2A)、同源异型盒基因D8(homeobox gene D8,HOXD8)、同源异型盒基因A9(homeobox gene A9,HOXA9)、肝细胞核因子4α(hepatocyte nuclear factor 4-alpha,HNF4A)、兔抗人受体相关孤儿受体α(rabbit anti-human retinoic acid receptor related orphan receptor A,RORA)(图3),表明这些转录因子与DEMis有调节关系。
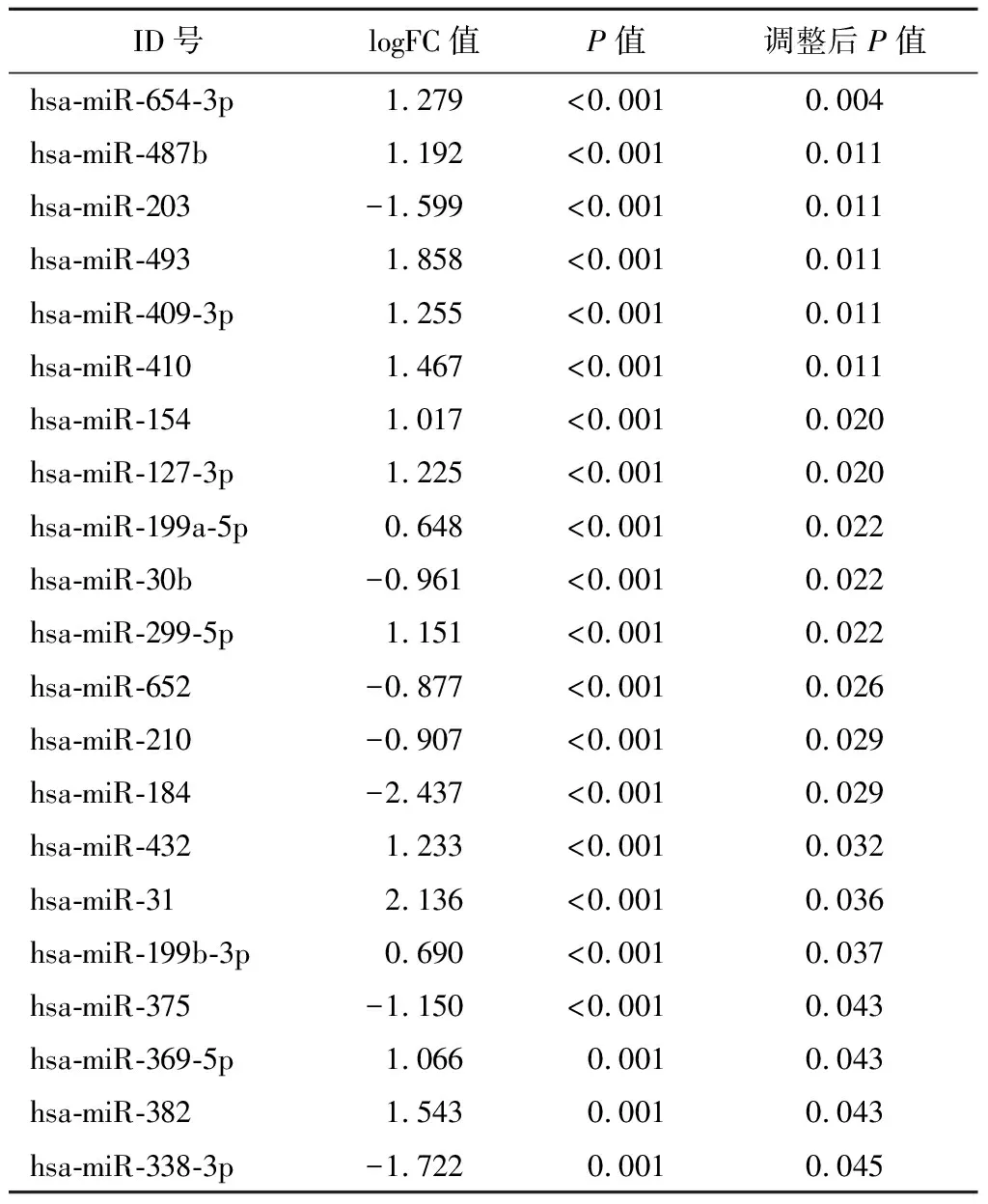
表1 IPF样本中21种差异表达的miRNA(DEMis)Table 1 21 differentially expressed miRNAs(DEMis) in IPF samples
2.3IPF中的DEMs筛选结果 本研究探讨了9例IPF患者和9例健康对照的mRNA表达水平。 mRNA的log(FC)阈值为0.47;确认了56个上调的mRNA和127个下调的mRNA。上调和下调的前20个基因(P<0.05为差异有统计学意义)见表2。火山图和热图见图4~5。
2.4在IPF中重建miRNA-mRNA网络 构建了一个带有3个mRNA和3个miRNA的miRNA-mRNA调控网络(图6),以进一步展示DEMis和DEMs之间的相互作用,有助于理解miRNA在IPF中的作用。网络中的度数、紧密度和中间度参数由Cytoscape软件(版本3.7.2)中的cytoHubba插件计算。调控网络显示miR-338-3p、miR-199a-5p和miR-299-5p可能作为关键miRNA在IPF发生和发展中发挥关键作用。跨膜6 超家族成员 1(transmembrane 6 superfamily member 1,TM6SF1)是miR-388-3p的靶标mRNA;轴突生长诱向因子G1(Netrin G1,NTNG1)是miR-199a-5p的靶标mRNA;钠通道电压门控Ⅱ型α(sodium channel voltage-gated type Ⅱ alpha,SCN2A)是miR-299-5p靶标mRNA。这3条途径可能与IPF的发展有关。
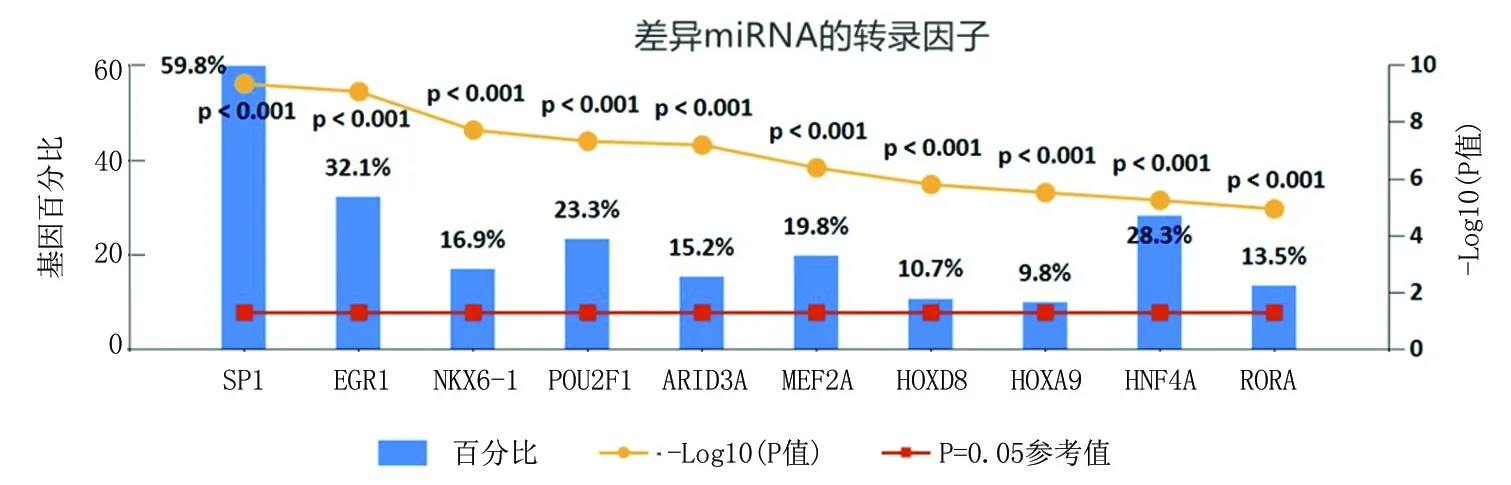
图3 DEMis的转录因子富集

图4 DEMs的火山图。红色:上调的基因;绿点:下调的基因

图5 DEM的热图。红色:高表达;绿色:低表达
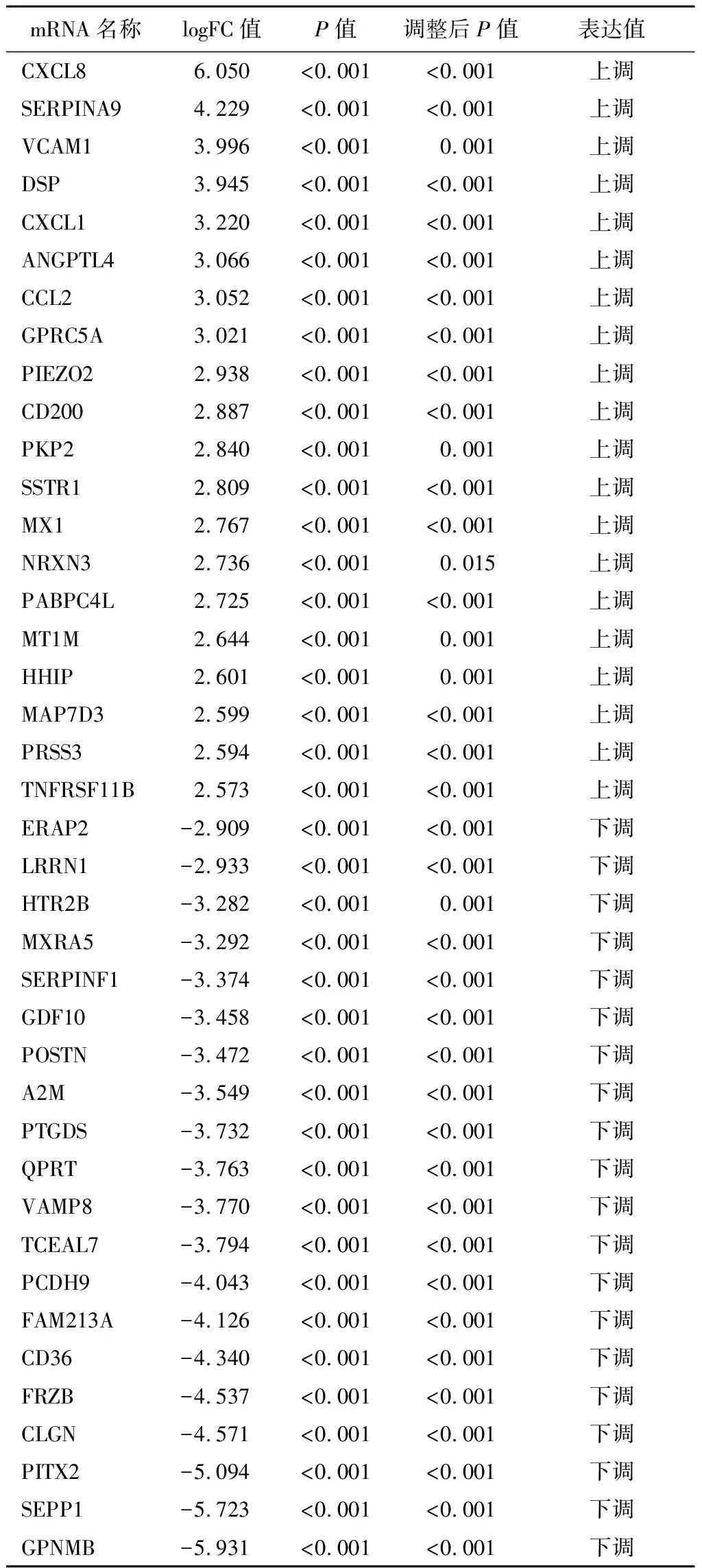
表2 前40个差异表达的mRNATable 2 The first 40 differentially expressed mRNAs
2.5调节网络中mRNA的功能富集分析 对调节网络中mRNA的GO分析表明,CC terms在电压门控钠通道复合物、突触膜的锚固成分、轴突旁节区、突触前、突触膜、谷氨酸能突触、突触膜的固有成分、突触前膜、突触前膜的固有成分、郎飞氏结等部位显著富集。分子功能途径包括细胞间黏附介体活性、钠通道活性、电压门控钠通道活性及细胞内黏附介体活性(图7),未做出BP途径富集。mRNAs与富集途径之间的关系见图8。KEGG最重要的3条路径(图9)包括味觉转导、细胞黏附分子及轴突导引。mRNA与KEGG富集途径的关系见图10。

图6 miRNA-mRNA网络。红色:上调的基因;绿色:下调的基因;mRNA用椭圆形表示,miRNA用三角形表示
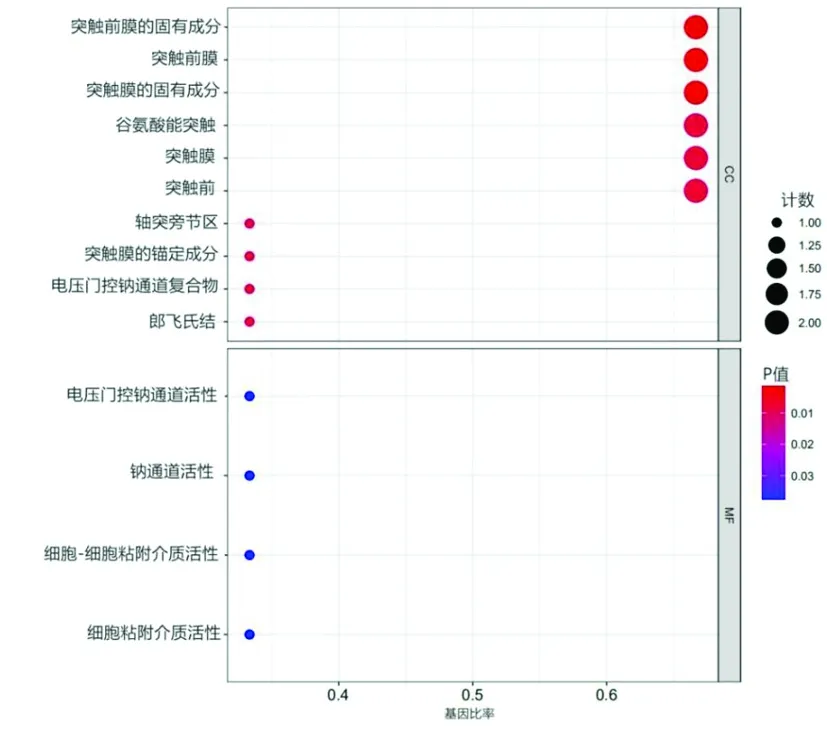
图7 IPF中DEMs富集的GO生物过程

图8 mRNAs与富集途径之间的关系

图9 网络中mRNA的KEGG途径富集分析

图10 KEGG中富集的mRNA之间的关系途径
3 讨 论
IPF是一种慢性纤维化性肺疾病,其特征是成纤维细胞的增殖和活化增加,包括成纤维细胞的积累、胶原蛋白的合成、细胞外基质蛋白和糖蛋白的沉积。目前该病无法治愈,具有复杂的临床表现,已批准吡非尼酮和尼达尼布用于减缓IPF的进展。miRNA是一类非编码的小RNA,其长度约为22个核苷酸,是基因调控中的重要调控因子。先前的研究表明,肺纤维化的发病机制与多种因素有关,包括DEMis和miRNA控制的差异基因表达[8]。因此,筛选和鉴定在IPF中差异表达的miRNA和基因,以及研究DEMis和DEMs之间的相关性,有助于阐明IPF发病的分子机制,并为临床新药的靶点研发提供指导。
GEO是基于R编程语言的分析工具,用于研究DEMs。本研究分析来自IPF和正常组织样品(GSE27430和GSE135065)的miRNA表达微阵列数据,并鉴定了21种差异表达的miRNA。这些miRNA中有7个下调,14个上调。在21种已鉴定的DEMis和183种常见DEMs调控的前20条信号通路中,至少有3条与IPF的进展有关,表明21个DEMis可能与IPF的进展有关。为了更好地了解DEMis靶标mRNA的机制,本研究筛选了可能的转录因子。其中SP1是最常见的转录因子,SP1参与调控与细胞外基质生成和降解密切相关基因(MMP14、PAI等)的表达,而细胞外基质过度沉积是肺纤维化的重要特征[9]。第2个主要的转录因子是EGR1,EGR1是肺纤维化常见的激活转录因子,可与SP1竞争结合MMP14启动子的GC序列,促进其转录表达[10]。而NKX6-1、POU2F1、ARID3A、MEF2A、HOXD8、HOXA9、HNF4A、RORA等转录因子在肺纤维化的发病机制中亦有不同的作用。由此可见,参与IPF的转录因子及其信号传导途径非常复杂,各种转录因子间会形成繁杂的信号网络系统,参与调控IPF相关基因的表达。
在肺上皮细胞和成纤维细胞中,miRNA是约22个碱基的小型非编码RNA,其可以通过靶向TGF-β信号传导事件,α平滑肌肌动蛋白和胶原蛋白,Ⅰ型基因表达影响纤维化活性,由此引发各种信号传导途径和生物进展,包括Smad介导和非氨介导的途径,分化、增殖、迁移、上皮间充质转换和细胞外基质[11-12]。迄今为止,已在人类中鉴定出约2 000种miRNA。有研究显示,在博来霉素诱导的肺纤维化小鼠模型中miR-199a-5p过度表达。然后在IPF患者的肺样品中,也通过在成纤维细胞灶内原位杂交,证实了其过表达[13]。表明miR-199a-5p可能与这种疾病的病理生理学有关,特别是在肺成纤维细胞的活化及其分化为成肌纤维细胞方面。实际上,miR-199a-5p在肺成纤维细胞中的过表达诱导了这些细胞的增殖、迁移和侵袭能力的增加,以及分化成肌纤维母细胞的能力。有研究分析了IPF肺中miRNA的表达,并通过定量RT-PCR验证了miR-299-5p的表达增加,并确定其在胚胎肺中类似地表达[14]。目前未检索到miR-388-3p在IPF发展中的作用,值得进一步探索。NTNG1和SCN2A是miRNA-mRNA网络中的关键基因。NTNG1是一种轴突导向分子,包含层粘连蛋白型EGF样结构域,并通过GPI锚点与质膜结合[15]。SCN2A基因位于染色体2q24.3,包含26个外显子,编码2 005个氨基酸。SCN2A基因编码电压门控钠离子通道α2亚基,主要分布在兴奋性神经元轴突起始段、未髓鞘化轴突部位,在大脑中分布广泛。到目前为止,与NTNG1相关的证据集中在精神和神经疾病上,与SCN2A相关的证据多集中在儿童癫痫[16],而两者与IPF之间的关联尚不清楚,是一个值得探索的新方向。
生物学过程和信号传导途径的富集分析表明,本研究中183个差异表达的基因与一系列生物学过程显著相关,例如细胞黏附和生物学黏附。ECM受体相互作用和细胞黏附是DEGs丰富的重要途径,已被证实与IPF的调节密切相关[17-18]。这些发现表明,183个差异表达基因在IPF基因表达谱数据集中呈显著差异表达,并且可能通过参与细胞黏附,生物黏附,ECM-受体相互作用等过程而参与IPF的进展。
本研究有一些局限性。首先应进一步解释IPF的潜在机制,并建立基因,miRNA,转录因子和mRNA组成的多重网络。此外,由于缺乏实验验证,需要进一步探索这些结论。
猜你喜欢
中国心脏起搏与心电生理杂志(2022年3期)2022-11-23
今日农业(2022年4期)2022-11-16
家庭医学(2022年5期)2022-04-27
火力与指挥控制(2022年3期)2022-04-27
福建中医药(2022年2期)2022-03-24
井冈山大学学报(自然科学版)(2022年1期)2022-02-28
昆明医科大学报(2019年3期)2019-10-21
师道·教研(2019年7期)2019-08-13
农家科技下旬刊(2016年8期)2017-05-05
中国民族民间医药·下半月(2011年10期)2011-12-27
