
有氧和厌氧环境下指示微生物的稳定性特征
2022-03-29 12:48郑前兴吴仁人李国栋陈中颖韦思业李开明
中国环境科学 2022年3期
张 杨,郑前兴,2,吴仁人*,李国栋,钟 义**,陈中颖,韦思业,李开明
有氧和厌氧环境下指示微生物的稳定性特征
张 杨1,郑前兴1,2,吴仁人1*,李国栋1,钟 义1**,陈中颖1,韦思业1,李开明1
(1.生态环境部华南环境科学研究所,广东 广州 510655;2.昆明理工大学环境科学与工程学院,云南 昆明 650500)
建立有氧和厌氧水环境模拟反应器,利用荧光定量PCR和高通量测序技术探究了猪源拟杆菌标记物、部分指示微生物和潜在致病菌在有氧及厌氧水环境中的变化特征.结果表明,指示微生物Streptococcaceae、Lactobacillaceae、Carnobacteriaceae、Ruminococcaceae、Lachnospiraceae和Prevotellaceae在不同反应器中均呈现出衰减趋势,但在有氧环境中的衰减幅度显著大于厌氧环境.因此,这几类指示微生物在有氧条件下适用于解析近期发生的污染事件.拟杆菌标记物GenBac和Pig-2-Bac在有氧环境和厌氧环境中均表现出二阶段衰减模式,且第一阶段为主要衰减阶段.但拟杆菌标记物在有氧环境中的衰减速率显著大于厌氧环境,表明有氧环境中拟杆菌标记物的稳定性较差.相关性分析显示潜在致病菌、和与拟杆菌标记物表现出较好的相关性(<0.05),但仅与标记物在有氧和厌氧环境中的相关性系数较为接近(0.913~0.953),表明拟杆菌标记物可作为的指示标记物.
粪便污染;微生物污染源解析;标记物;指示微生物;稳定性
随着我国畜禽养殖业的飞速发展,粪便废水排放已逐渐成为地表水水质恶化的主要原因之一,粪便中携带的致病微生物还能导致介水性疾病的爆发,给人类的健康带来潜在威胁[1-2].为有效防治粪便废水的污染,粪大肠菌群、大肠埃希氏菌和肠球菌等传统指示微生物一直作为判断水体受粪便污染程度的指标.然而,传统指示微生物可能在温度条件适宜,营养水平较高的水体中天然存在[3-4],且无法提供粪便污染宿主来源等信息[5].微生物污染源解析技术(MST)可以通过分子生物学手段识别污染物的宿主来源,还可以排除自然环境中土著微生物对源解析结果的干扰[6-7].MST技术通常可分为非库依赖法和库依赖法两类.非库依赖法主要是针对不同宿主肠道内特征微生物的基因片段开发出相应的标记物,具有易操作、成本低、时效强等优势[5].
目前,大多数标记物主要针对各宿主肠道内的拟杆菌特异性基因片段开发而来.一方面,拟杆菌标记物具有较强的宿主特异性,如猪源拟杆菌标记物Pig-1-Bac和Pig-2-Bac在我国有较好的适用性,特别是Pig-2-Bac的特异性达到了88%,可有效识别来自于猪粪废水的污染[8].另一方面,拟杆菌作为专性厌氧菌,在进入第二生境后难以增殖,不会影响对于粪便污染水平的判断.而传统指示微生物,如大肠埃希氏菌可在适宜的环境中增殖,使得无法准确评估环境中的粪便污染强度[9-10].库依赖法是利用高通量测序技术分析不同宿主粪便中的微生物信息,建立粪便污染分类文库,进而与自然环境中获取的微生物信息进行对比,识别粪便排放的宿主来源[11].该方法的优势在于不仅可以提供污染源信息,还能提供粪便及自然水体中微生物群落结构和致病菌分布等情况,但该方法存在难以进行准确定量分析的缺陷[12].因此,目前MST逐渐发展为同时结合2种技术方法,充分分析各污染源的污染强度及样本中的微生物群落信息.
尽管MST技术相比于传统指示微生物具有诸多优势,但要准确评估各污染源的贡献率还需要掌握标记物或肠道微生物在不同水环境中的稳定性特征.目前已有研究探索了不同温度、营养水平条件下标记物在水环境中的变化规律[9,13],但关于溶解氧对标记物或肠道微生物环境行为的影响研究相对较少.在黑臭水体或富营养化较为严重的水环境中,由于大量有机污染物的存在和微生物降解等作用[14],水体可能呈现厌氧状态,而溶解氧的变化对于微生物群落组成有显著影响[15-16],因此在有氧和厌氧水环境中,标记物或肠道微生物的稳定性表现可能会有较大差异,从而影响源解析的准确性.此外,由于致病菌在环境中的丰度普遍较低,因此研究人员一直致力于寻找可靠的指示微生物用于评价水质污染和健康风险[17-19].已有研究报道了人源拟杆菌标记物HF183和Hum2对于潜在致病菌的指示作用[9,20].然而,拟杆菌标记物与潜在致病菌之间的相关关系可能会因宿主不同而发生变化[21],目前关于猪粪携带的潜在致病菌与猪源拟杆菌标记物的相关关系研究较少.本研究通过搭建的水环境模拟反应器,模拟猪源肠道微生物在有氧和厌氧水环境中的变化情况,探究溶解氧对于标记物和肠道微生物稳定性的影响,以期为高效开展微生物污染源解析工作提供参考.
1 材料与方法
1.1 样品及试剂
样品采集:猪粪样品取自广东省某畜牧养殖研究所.利用灭菌勺和灭菌塑料瓶采集新鲜猪粪并放入冰盒中保存,2h内运回实验室进行处理.
主要仪器:荧光定量PCR仪(Light Cycler 480II, Roche公司),紫外可见分光光度计(DR2800,HACH公司),磁力搅拌器(上海司乐仪器公司), 溶解氧测定仪(Pro20,YSI 公司),冷冻离心机(Sigma 公司).
主要试剂:水体DNA提取试剂盒(D3145-02, Magen公司),荧光染料SYBR TaqⅡ(TIANGEN公司), COD 高量程预制试剂(HACH公司), LB肉汤培养基(环凯生物公司),琼脂糖(Biowest,电泳级).
1.2 模拟反应器搭建
水环境模拟反应器采用透明有机玻璃制成,反应器为圆柱形,高25cm,直径为30cm.反应器可置于磁力搅拌器上,通过长度为8cm的转子对反应器内的水体进行搅拌,转速约为170~220r/min.
实验分为有氧组(AeT)和厌氧组(AnT),每组设置2个反应器作为平行实验,共建立4个反应器.为排除土著微生物对反应器内粪源微生物的干扰,有效识别有氧和厌氧环境下标记物及指示微生物的稳定性差异,实验采用粪便接种至去离子水的方式构建各反应器.反应器建立之前,采用4L灭菌去离子水溶解8g猪粪样本,充分混匀后依次在37,29μm灭菌滤网中过滤以去除可能干扰实验的杂质.过滤完成后,对制备的粪液进行充分混匀,并均分为4份分别置于4个反应器当中,每个反应器均通过灭菌去离子水定容至12L.反应器搭建完成后,向厌氧组的2个反应器内通入N2使得反应器中水体的溶解氧含量降至0.1mg/L以下,并在整个实验过程中保持25L/h的通气量,维持厌氧组反应器的厌氧环境.有氧和厌氧组反应器均置于磁力搅拌器上,通过转子对水体进行持续搅拌.当有氧组中拟杆菌标记物的浓度趋于稳定,不发生显著变化时,同时结束两组实验,对标记物的稳定性特征进行分析,实验共持续11d.实验运行期间,有氧和厌氧反应器温度变化较为相似,均在18~23℃.有氧反应器的pH值变化范围在6.61~7.78,小于厌氧反应器的pH值变化范围(7.79~9.05).
1.3 分析方法
1.3.1 DNA提取和测定 每日从各反应器中采集100mL水样,开展微生物的变化检测.水样采集后采用0.22μm滤膜对其进行抽滤,将微生物截留于滤膜上,按照水体DNA提取试剂盒说明书提取截留在滤膜上的DNA,放入-20℃冰箱留待qPCR和高通量测序分析.
1.3.2 qPCR和高通量测序 选取拟杆菌通用引物(GenBac)、大肠埃希氏菌引物(EC23S857)和猪源特异性拟杆菌引物(Pig-2-Bac)分析不同水环境中标记物的变化特征,引物信息如表1所示.
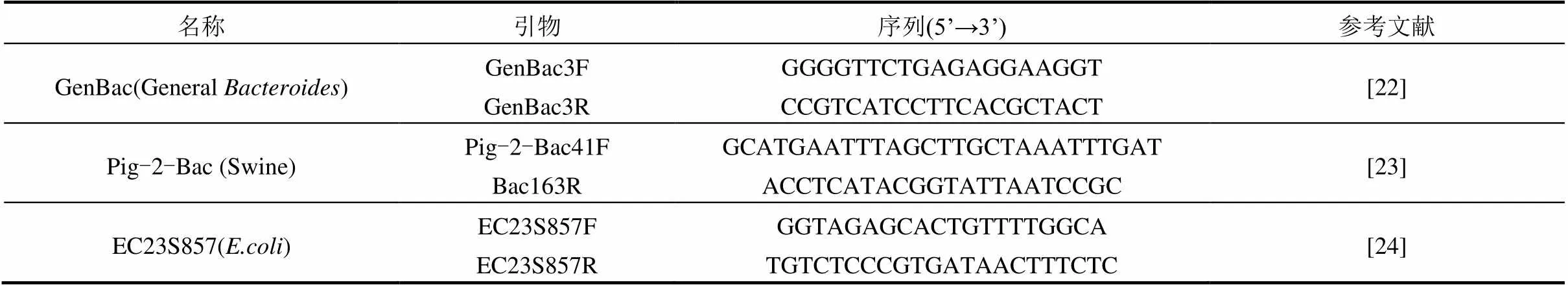
表1 标记物信息
qPCR扩增体系为20μL:DNA模板2μL; SYBR TaqⅡ 10μL;10μmol/L正、反向引物各0.8μL;ddH2O补足体系至20μL.扩增程序采用两步法: ① 95℃预变性30min;② 95℃变性5s;③60℃退火延伸15s,重复40个循环.荧光染料qPCR的特异性通过熔解曲线分析实现.
标准曲线:采用已提取的DNA为模板,通过普通PCR进行扩增,对扩增后的产物纯化回收,并连接到PMDTM19-T载体上,转化至DH5α感受态细胞中,通过平板划线法挑取阳性克隆,提取质粒.通过超微量分光光度计测定质粒浓度,计算目的片段拷贝数.同时对质粒进行10倍梯度稀释,分别稀释成10-1,10-2,10-3,10-4,10-5,10-66个梯度,每个梯度的质粒作为标准品设置3个重复,进行qPCR扩增.反应结束后制作标准曲线.标准曲线的相关系数均大于0.99,扩增效率为92%~102%.
16S rDNA高通量测序由广州基迪奥测序公司完成.针对已提取的DNA,选取基因的V3~V4区进行PCR扩增,采用的引物为341F (5-CCTAYGGG- RBGCASCAG-3)和806R(5-GGACTACNNGGGT- ATCTAAT-3).通过凝胶回收试剂盒(TIANGEN公司)切胶回收PCR产物后,利用QuantiFluorTM荧光计系进行定量.按Illumina HiSeq平台测序要求开展测序.高通量测序数据的序列上传至NCBI序列读取档案,登记号为SUB4479220.原始序列经质控后获得高质量序列,并在SILVA数据库进行比对分析.
1.3.3 数据分析 标记物衰减规律:根据标记物在水体中的变化特征,分别采用一阶和二阶衰减模型对标记物的变化进行拟合,获取衰减速率[25-27].当标记物的衰减规律存在明显拐点,则采用二阶衰减模型,否则采用一阶衰减模型[25].
对于表现出二阶段衰减趋势的标记物,其浓度下降主要发生在第1阶段,因此由二阶衰减模型模拟的标记物衰减过程,90计算时仅考虑第1阶段衰减速率(1).
相关性分析:为探索潜在致病菌与猪源拟杆菌标记物的相关关系,采用SPSS软件进行Pearson相关性统计分析.
2 结果与讨论
2.1 标记物在有氧/厌氧水环境中的衰减规律
由图1可以看出,在实验结束时,选用的3个标记物在厌氧水环境中的浓度下降了约2个数量级,而在有氧环境中则下降了3~4个数量级,衰减幅度显著高于厌氧环境.衰减趋势方面,由AIC(赤池信息量准则)分析结果可知GenBac和Pig-2-Bac在所有水环境模拟反应器中的浓度变化均表现出二阶段衰减模式,存在拐点(图1a,b).二阶段衰减模型的模拟结果显示(表2),GenBac在有氧和厌氧环境中的拐点分别为第6.19d和第10.25d,Pig-2-Bac在有氧和厌氧环境中的拐点分别为7.14和9.76d,表明拟杆菌标记物在有氧水环境中的第1衰减阶段耗时普遍小于厌氧水环境,在溶解氧作用下,拟杆菌标记物经历了剧烈变化过程以适应环境,这也说明溶解氧对于拟杆菌标记物的稳定性影响显著.EC23S857在有氧环境中表现出2阶段衰减趋势,浓度变化的拐点为第8d,但在厌氧环境中则近似于线性衰减模型.由AIC结果也可知,一阶衰减模型对于EC23S857在厌氧环境中浓度变化的模拟效果优于二阶衰减模型.
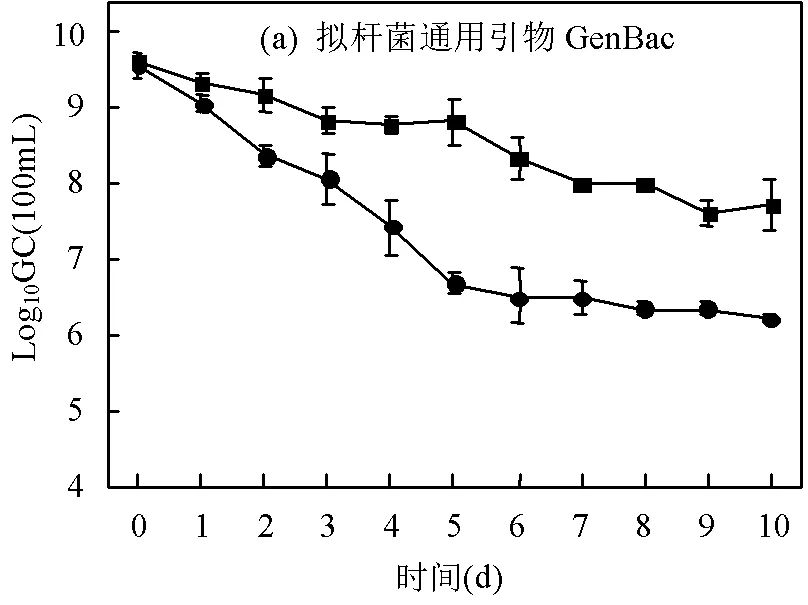
表2可以看出,拟杆菌标记物GenBac和Pig-2- Bac在有氧环境下的90小于厌氧环境3~5d,2种不同模拟环境下GenBac和Pig-2-Bac的浓度变化规律也具有显著差异(<0.05),表明有氧和厌氧条件对于拟杆菌标记物的环境行为具有显著影响.导致这一现象产生的原因主要是拟杆菌标记物的第一阶段衰减速率(1)在有氧和厌氧条件下差异(<0.05)较大所致.GenBac和Pig-2-Bac在有氧环境中的1分别高于厌氧环境62%和66%,而不同模拟条件下标记物的第2阶段衰减速率(2)则差异较小.这表明拟杆菌在进入第二生境后,厌氧环境可有效避免溶解氧对拟杆菌的毒性作用,增加了标记物在水体中的稳定性,使其可以对时间跨度较长的污染事件及污染源进行追踪解析[28].
对比拟杆菌标记物2个阶段的衰减速率还可以看出,在不同水环境中标记物的1普遍高于2,说明标记物的浓度衰减主要发生在第一阶段.尽管已有诸多研究报道了光照、温度、营养水平及土著微生物等外界因素对于拟杆菌标记物衰减的作用机制各不相同,但衰减规律普遍呈现出2阶段趋势,且第1阶段为主要衰减阶段,而本研究从溶解氧的角度也进一步揭示了二阶段衰减模型普遍适用于模拟拟杆菌标记物的变化规律[26,29].目前,关于导致标记物两阶段衰减速率差异较大的成因尚不明确.Easton等推测可能是由于此类微生物在进入水环境后,逐渐适应了环境压力,导致出现了衰减速率较低的第二衰减阶段[30-31].Strauss等[32]也认为可能是微生物群落通过群体感应适应了环境压力,大幅减小了衰减速率.但具体原因有待进一步研究.
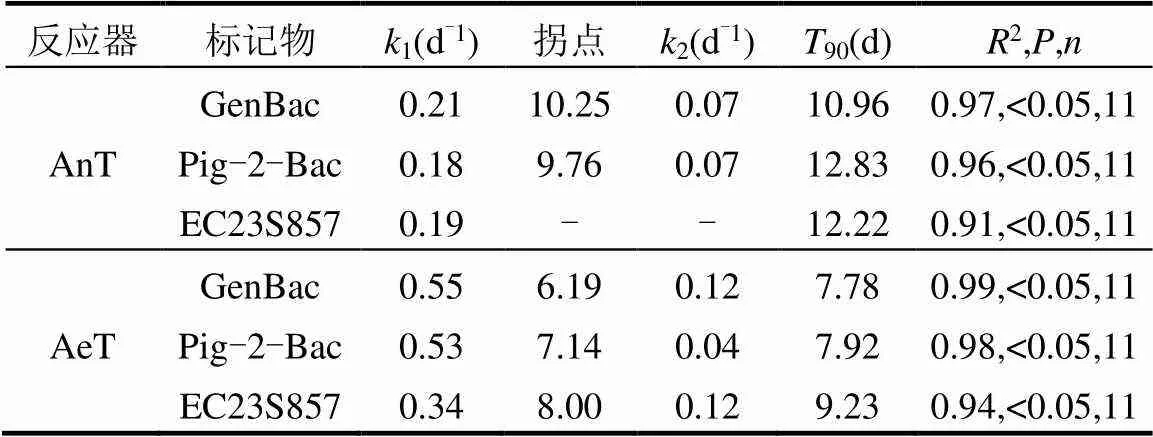
表2 标记物在有氧/厌氧水环境中的衰减速率及衰减周期
注:1:一阶段衰减速率;2:二阶段衰减速率;:显著性;:实验天数;“-”代表一阶衰减模拟不存在拐点和2.
相比于拟杆菌标记物,有氧和厌氧环境对于标记物的影响较小(图1c).尽管EC23S857在有氧和厌氧模拟反应器中表现出了不同的衰减趋势,但其在厌氧环境中的衰减速率与有氧环境中第一衰减阶段的衰减速率差异较小,同时二者的浓度变化也未表现出显著性差异(>0.05).这与Pachepsky等[33-34]对于在水体中变化的研究结果一致,即作为兼性厌氧微生物,其对溶解氧变化的响应远小于光照和浊度等其它环境因素.
2.2 微生物群落分布
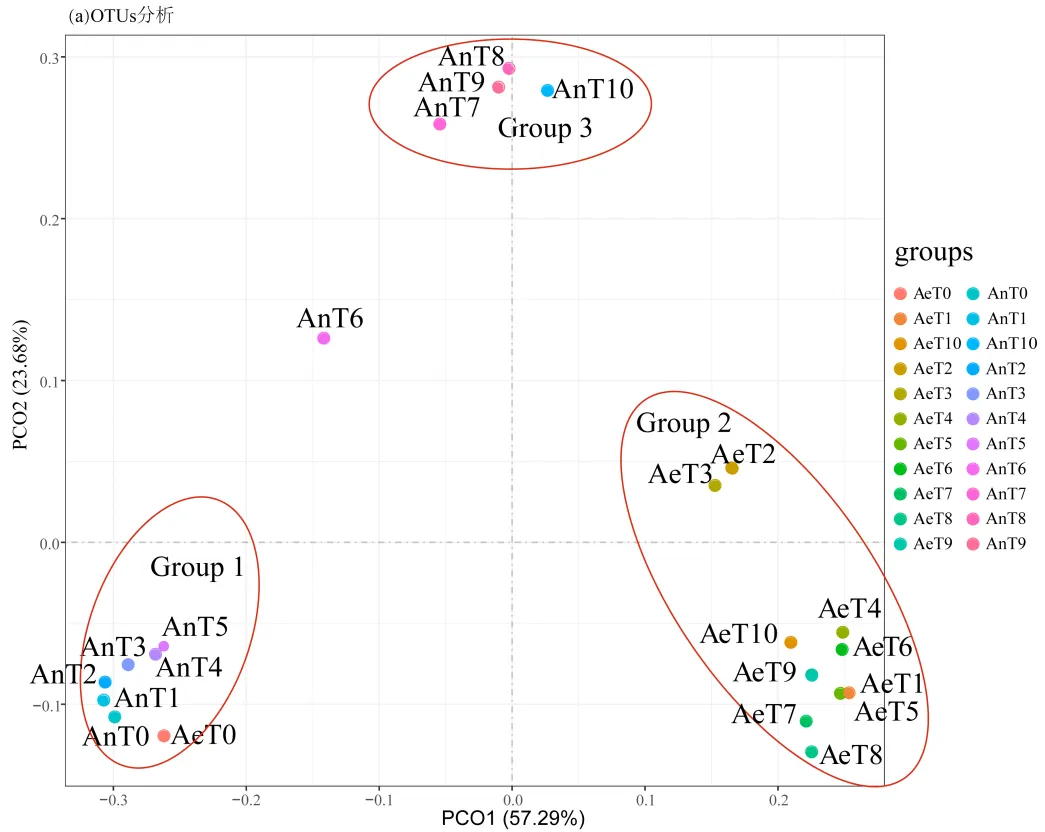
对好氧和厌氧反应器中所有采集的样本进行门水平微生物群落结构分析(图2b),结果显示Firmicutes(厚壁菌门)在样本中占主要地位,平均相对丰度为40.3%,其次为Proteobacteria(变形菌门, 25.7%),Bacteroidetes(拟杆菌门,22.7%), Actinobacteria(放线菌门,5.9%),Verrucomicrobia(疣微菌门,4.6%)和Planctomycetes(浮霉菌门,0.2%),该结果与较多研究一致,表明Firmicutes、Proteobacteria和Bacteroidetes是猪肠道中的优势菌群[35-36].采用PCA分析样本中的OTUs种类发现,所有样本的OTUs种类可分为3组(图2a),第1组主要包括AeT0(有氧反应器第0d样本)和AnT0~AnT5,该组中Firmicutes占主要地位.第2组中包含了大部分有氧反应器中的样本,该组中Bacteroidetes占主要地位.第3组中主要包括AnT7~AnT10, Proteobacteria占主要地位.在3个分类组之间,Actinobacteria、Planctomycetes的相对丰度并未表现出显著差异(T检验,>0.05).
2.3 指示微生物的变化规律
在科水平上考察有氧和厌氧反应器中水样的微生物组成和变化规律(图3).相对丰度大于1%的肠道微生物共有13类,其中相对丰度最高的为Comamonadaceae(13.5%),其次为Streptococcaceae (8.3%), Moraxellaceae(7.1%), Flavobacteriaceae (6.7%), Cryomorphaceae(6.2%), Lactobacillaceae (5.8%), Clostridiaceae(5.6%), Verrucomicrobiaceae (4.6%), Peptostreptococcaceae(4.0%), Carnobacteriaceae (3.9%), Ruminococcaceae(3.3%), Lachnospiraceae (2.3%)和Prevotellaceae(2.2%).在13类微生物当中, Streptococcaceae、Lactobacillaceae、Carnobacteriaceae、Ruminococcaceae、Lachnospiraceae和Prevotellaceae 6类微生物在有氧和厌氧反应器中的相对丰度均表现出下降的趋势,而其它微生物则发生增殖或波动的现象,说明6类指示微生物的变化规律较为简单且易于预测,符合作为指示微生物的特征[12,37].而已有部分研究也指出,上述6类指示微生物除具有一定宿主指示功能外,还因其在环境中的变化规律较为单一而常被视作有效的指示微生物用于库依赖法源解析工作中[11-12,21].
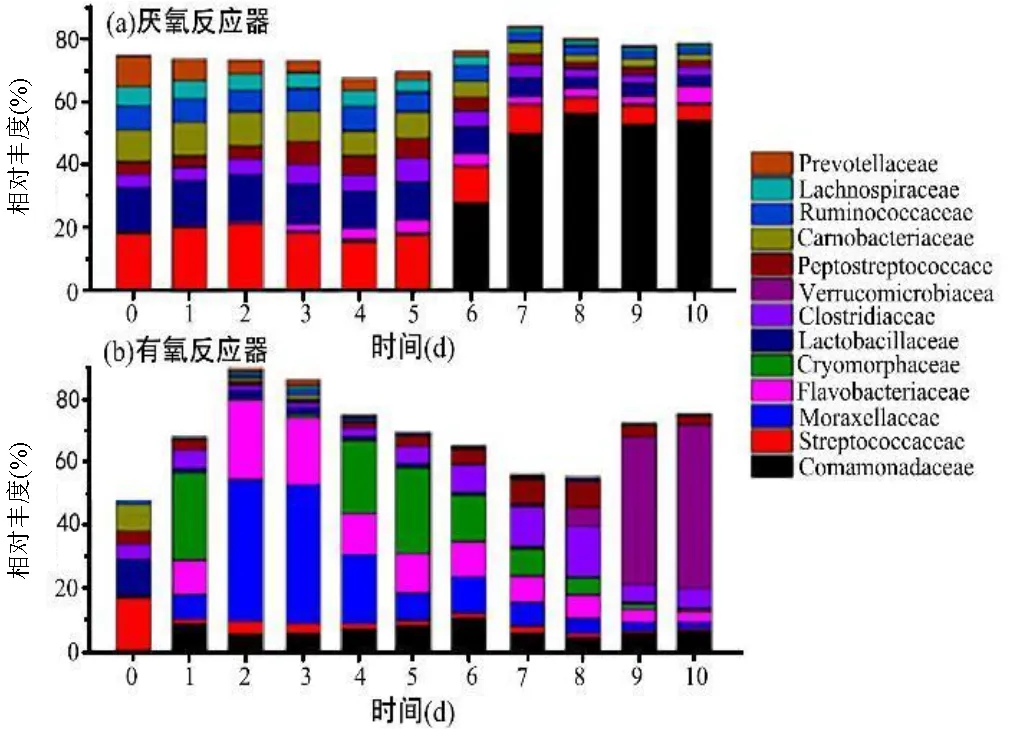
图3 猪源肠道微生物在反应器中相对丰度的变化
然而,随着实验的进行,6类指示微生物在厌氧反应器中的衰减速率显著小于有氧反应器.厌氧环境中,除Prevotellaceae在实验结束时的相对丰度较初始情况下降了约25倍,其它5类指示微生物的终末相对丰度较初始情况仅下降了约2~6倍(图 3a).而有氧环境中,6类指示微生物相对丰度的衰减程度显著大于厌氧环境,特别是在实验开始的前2d,6类指示微生物的相对丰度在整个微生物群落中的占比迅速下降至1%以下,随后保持较低丰度直至实验结束(图3b).由上述实验结果可以看出,厌氧环境可以使得指示微生物在一定程度上免于溶解氧带来的毒害或抑制作用,指示微生物在该环境条件下,例如黑臭水体或沉积物当中,能够维持较好的稳定性,可用于追溯污染事件发生时间相对较久的污染源.有氧环境中,指示微生物会在进入水体初期发生快速衰减,可能导致低估污染源的污染水平,因此仅能用于解析近期污染事件中的污染源.而随后在第二衰减阶段维持较低水平,则可能是由于指示微生物逐渐适应了环境压力,同时环境中的营养物质已足够支持低丰度指示微生物的生活方式所致[30-31].
2.4 潜在致病菌的分布特征及变化规律
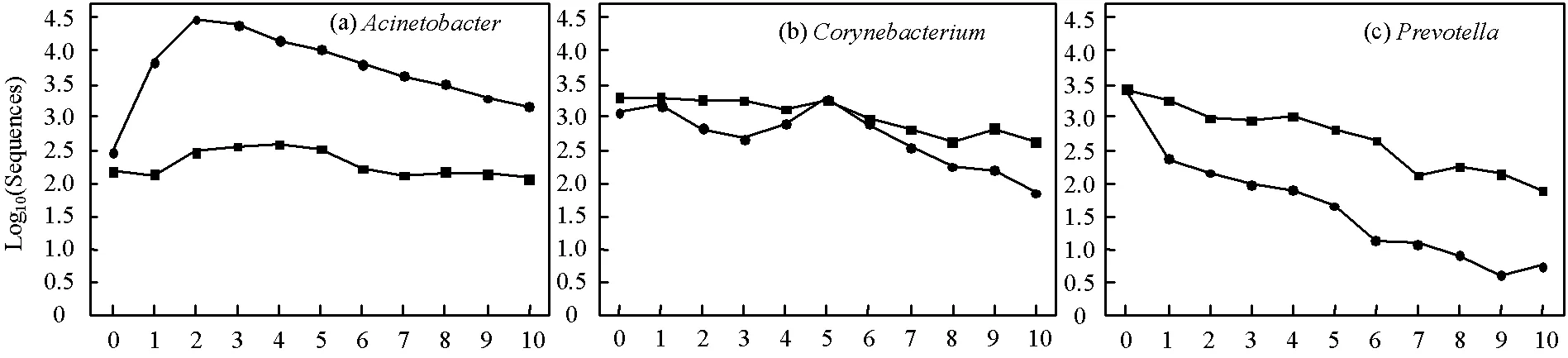
为探究粪源致病菌在有氧和厌氧环境中的变化规律,本研究参照以往关于粪源致病菌分布的研究,在属水平识别出相对丰度大于0.1%的潜在致病菌共6类[21,38-39].总体而言,的相对丰度最高,达到了72.0%,其次为(14.6%)、(8.3%)、(2.8%)、(1.5%)和(0.4%),其中(图4f)仅在厌氧反应器中检出.整个实验过程中,(图4b)、(图4d)和(图4e)在厌氧环境中的相对丰度呈现缓慢下降趋势,但衰减过程中存在波动变化的现象,特别是和,相对丰度在实验的第1~3d有所回升,这可能是微生物对环境变化存在一定的应激响应过程[40-41].(图4a)在有氧环境第2d的相对丰度较初始值增长了约2个数量级,随后呈现出缓慢下降的趋势,但在厌氧环境中的相对丰度则变化较小.这与已有关于的研究较为一致,即作为可引起呼吸道感染及脑膜炎等疾病的一类致病菌,通常为专性需氧菌,在有氧条件下,可利用常见营养物质进行繁殖[42].然而,近年来也有研究人员通过厌氧培养方式获得了的部分菌株,说明厌氧环境中部分菌株可能也具有一定的活性[43],这可能也是本研究中未出现明显衰减趋势的原因之一,但具体原因仍需进一步分析.在识别的6类潜在致病微生物当中,仅(图4c)在有氧和厌氧环境中均表现出衰减趋势,但其在有氧环境中的衰减幅度大于厌氧环境约1.5个数量级,表明溶解氧对于的稳定性有显著影响.
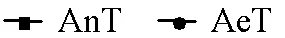
2.5 潜在致病菌与标记物的相关性
本研究识别的相对丰度大于0.1%的6类潜在致病菌当中,仅、和等3类微生物与猪源拟杆菌标记物在有氧和厌氧两种环境中均表现出较好的相关性(<0.05)(表3).值得注意的是,与标记物在有氧和厌氧环境中的相关性系数较为接近(0.913~0.953),表明拟杆菌标记物与在不同环境中的变化规律较为相似,可作为的指示物用于健康风险评估.和与拟杆菌标记物在厌氧环境中的相关性均高于有氧环境,这可能是因为和作为兼性菌,在有氧环境中的衰减存在一定的波动性,导致其衰减规律与拟杆菌标记物的二阶段衰减模式存在一定的差异性.已有研究报道了对于需氧或兼性厌氧的潜在致病菌而言,即使在有氧环境中短暂的增殖也会影响与拟杆菌标记物之间的相关关系,进而干扰拟杆菌标记物对致病菌的指示作用[9].
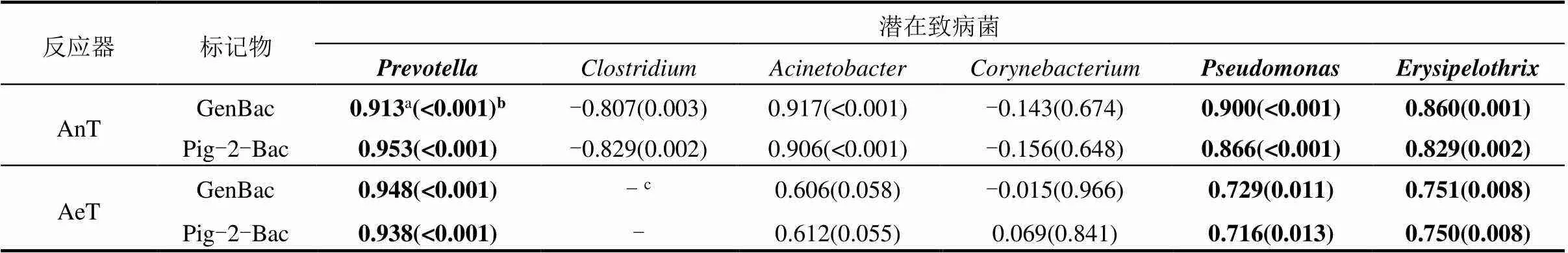
表3 潜在致病菌和标记物的相关性
注:a相关性系数;b在0.05级别相关性显著(<0.05);c“-”在AeT中未检出;黑体表示在2个反应器(AnT、AeT)中,均与标记物GenBac、Pig-2-Bac表现出显著相关性的潜在致病菌.
3 结论
3.1 拟杆菌标记物GenBac和Pig-2-Bac在有氧和厌氧环境中均表现出二阶段衰减模式,主要衰减过程发生在第一阶段.但标记物在有氧环境中的衰减速率显著大于厌氧环境,其在有氧环境中的稳定性较差.
3.2 识别出的6类指示微生物Streptococcaceae、Lactobacillaceae、Carnobacteriaceae、Ruminococcaceae、Lachnospiraceae和Prevotellaceae在有氧和厌氧环境中均表现出衰减趋势,可用于推测粪便污染水平.但6类指示微生物在有氧环境中的衰减幅度均大于厌氧环境,适用于解析有氧环境中的近期污染事件.
3.3 相对丰度大于0.1%的潜在致病菌当中,仅、和与拟杆菌标记物表现出较好的相关性(<0.05).但仅有与标记物在有氧和厌氧环境中的相关性系数较为接近(0.913~0.953),因此拟杆菌标记物可作为的指示物用于健康风险评估.
[1] Wéry N, Monteil C, Pourcher A M, et al. Human-specific fecal bacteria in wastewater treatment plant effluents [J]. Water Research, 2009,44(6):1873-1883.
[2] Wu B L, Wang X C, Dzakpasu M. Genetic characterization of fecal impacts of seagull migration on an urban scenery lake [J]. Water Research, 2017,117:27-36.
[3] Emma R N, Nguyen T M H, Le T P Q, et al. A short review of fecal indicator bacteria in tropical aquatic ecosystems: knowledge gaps and future directions [J]. Frontiers in Microbiology, 2015,6:308.
[4] Winfield M D, Groisman E A. Role of nonhost environments in the lifestyles ofand[J]. Applied and Environmental Microbiology, 2003,69(7):3687-3694.
[5] Ahmed W, Gyawali P, Feng S, et al. Host-specificity and sensitivity of the established and novel sewage-associated marker genes in human and non-human fecal samples [J]. Applied and Environmental Microbiology, 2019,85(14):e00641-19.
[6] Hagedorn C, Blanch A R, Harwood V J. Microbial source tracking: methods, applications, and case Studies [M]. New York: Springer, NY, 2011:301-312.
[7] Harwood V J, Staley C, Badgley B D, et al. Microbial source tracking markers for detection of fecal contamination in environmental waters: relationships between pathogens and human health outcomes [J]. Fems Microbiology Reviews, 2014,38(1):1-40.
[8] Zhang Y, Wu R R, Lin K R, et al. Performance of host-associated genetic markers for microbial source tracking in China [J]. Water Research, 2020,175:115670.
[9] Zhang Y, Wu R R, Zhang Y M, et al. Impact of nutrient addition on diversity and fate of fecal bacteria [J]. Science of the Total Environment, 2018,636:717-726.
[10] Brooks Y M, Spirito C M, Bae J S, et al. Fecal indicator bacteria, fecal source tracking markers, and pathogens detected in two Hudson River tributaries [J]. Water Research, 2020,17:115342.
[11] Staley C, Kaiser T, Lobos A, et al. Application of SourceTracker for accurate identification of fecal pollution in recreational freshwater: a double-blinded study [J]. Environmental Science and Technology, 2018,52:4207-4217.
[12] Brown C M, Staley C, Wang P, et al. A high-throughput DNA- sequencing approach for determining sources of fecal bacteria in a lake Superior estuary [J]. Environmental Science and Technology, 2017,51(15):8263-8271.
[13] Warish A, Christopher S, Thomas K, et al. Decay of sewage- associated bacterial communities in fresh and marine environmental waters and sediment [J]. Applied Microbiology and Biotechnology, 2018,102:1-12.
[14] Rhodes J, Hetzenauer H, Frassl M A, et al. Long-term development of hypolimnetic oxygen depletion rates in the large Lake Constance [J]. Ambio., 2017,46:554-565.
[15] Marti R, Mieszkin S, Solecki O, et al. Effect of oxygen and temperature on the dynamic of the dominant bacterial populations of pig manure and on the persistence of pig-associated genetic markers, assessed in river water microcosms [J]. Journal of Applied Microbiology, 2011,111(5):1159-1175.
[16] Gao C, Wang A, Wu W M, et al. Enrichment of anodic biofilm inoculated with anaerobic or aerobic sludge in single chambered air- cathode microbial fuel cells [J]. Bioresource Technology, 2014,167: 124-132.
[17] Sokolova E, Aström J, Pettersson T, et al. Decay ofgenetic markers in relation to traditional fecal indicators for water quality modeling of drinking water sources [J]. Environmental Science and Technology, 2011,46(2):892-900.
[18] Griffin D W, Lipp E K, Mclaughlin M R, et al. Marine recreation and public health microbiology: quest for the ideal indicator [J]. BioScience, 2001,(10):817-825.
[19] Savichtcheva O, Okayama N, Okabe S. Relationships between16S rRNA genetic markers and presence of bacterial enteric pathogens and conventional fecal indicators [J]. Water Research, 2007,41(16):3615-3628.
[20] Boehm A B, Soller J A, Shanks O C. Human-associated fecal quantitative polymerase chain reaction measurements and simulated risk of gastrointestinal illness in recreational waters contaminated with raw sewage [J]. Environmental Science and Technology Letters, 2015,2(10):270-275.
[21] Nshimyimana J P, Freedman A, Shanahan P, et al. Variation of bacterial communities with water quality in an urban tropical catchment [J]. Environmental Science and Technology, 2017,51(10): 5591-5601.
[22] Siefring S, Varma M, Atikovic E, et al. Improved real-time PCR assays for the detection of fecal indicator bacteria in surface waters with different instrument and reagent systems [J]. Journal of Water and Health, 2008,6(2):225-237.
[23] Mieszkin S, Furet J P, Corthier G, et al. Estimation of pig fecal contamination in a river catchment by real-time PCR using two pig-specific16S rRNA genetic markers [J]. Applied and Environmental Microbiology, 2008,75(10):3045-3054.
[24] Chern E C, Siefring S, Paar J, et al. Comparison of quantitative PCR assays fortargeting ribosomal RNA and single copy genes [J]. Letters in Applied Microbiology, 2011,52(3):298–306.
[25] Ahmed W, Zhang Q, Kozak S, et al. Comparative decay of sewage-associated marker genes in beach water and sediment in a subtropical region [J]. Water Research, 2019,149:511-521.
[26] Jeanneau L, Solecki O, Wery N, et al. Relative decay of fecal indicator bacteria and human-associated markers: A microcosm study simulating wastewater input into seawater and freshwater [J]. Environmental Science and Technology, 2012,46(4):2375–2382.
[27] Zhang Y, Wu R R, Li W J, et al. Occurrence and distributions of human-associated markers in an impacted urban watershed [J]. Environmental Pollution, 2021,275(14):116654.
[28] Mantilla-Calderon D, Hong P Y. Fate and persistence of a pathogenic NDM-1-positivestrain in anaerobic and aerobic sludge microcosms [J]. Applied and Environmental Microbiology, 2017,83(13):e00640-17.
[29] Liu R L, Cheng K H F, Wong K, et al. Differential utility of theDNA and RNA markers in the tiered approach for microbial source tracking in subtropical seawater [J]. Applied Microbiology and Biotechnology, 2015,99(13):5669-5681.
[30] Easton J H, Gauthier J J, Lalor M M, et al. Die-soff pathogenicO157: H7 in sewage contaminated waters [J]. Jawra Journal of the American Water Resources Association, 2010,41(5):1187-1193.
[31] Rogers S W, Donnelly M, Peed L, et al. Decay of bacterial pathogens, fecal indicators, and real-time quantitative PCR genetic markers in manure-amended soils [J]. Applied and Environmental Microbiology, 2011,77(14):4839-4848.
[32] Strauss E. Microbes, immunity, and disease:a symphony of bacterial voices [J]. Science, 1999,284(5418):1302-1304.
[33] Pachepsky Y, Kierzewski R, Stocker M, et al.Temporal stability ofconcentrations in waters of two irrigation ponds in Maryland [J]. Applied and Environmental Microbiology, 2018,84(3): e01876-17.
[34] Dwivedi D, Mohanty B P, Lesikar B J. Impact of the linked surface water-soil water-groundwater system on transport ofin the subsurface [J]. Water Air and Soil Pollution, 2016,227(9):351.
[35] 王文文,王 园,安晓萍,等.断奶仔猪肠道菌群结构特征及其调控研究进展[J]. 动物营养学报, 2020,32(11):4998-5005.
Wang W W, Wang Y, An X P, et al. Research advances on structural characteristics and modulation of gut microbiota of weaned piglets [J]. Chinese Journal of Animal Nutrition, 2020,32(11):4998-5005.
[36] 郑 青.不同饲养模式对猪肠道菌群结构的影响研究[D]. 合肥:安徽农业大学, 2017.
Zheng Q. The effects of different breeding patterns on intestinal microflora structure in pigs [D]. Hefei: Anhui Agricultural University, 2017.
[37] Eichmiller J J, Borchert A J, Sadowsky M J, et al. Decay of genetic markers for fecal bacterial indicators and pathogens in sand from Lake Superior [J]. Water Research, 2014,59:99-111.
[38] Wattam A R, David A, Dalay O, et al. PATRIC, the bacterial bioinformatics database and analysis resource [J]. Nucleic Acids Research, 2014,42(D1):D581-D591.
[39] Cui Q J, Fang T T, Huang Y, et al. Evaluation of bacterial pathogen diversity, abundance and health risks in urban recreational water by amplicon next-generation sequencing and quantitative PCR. [J]. Journal of environmental sciences (China), 2017,57:137-149.
[40] Lee J Y, Kim H J, Kim E S, et al. Regulatory interaction of the Corynebacterium glutamicum whc genes in oxidative stress responses [J]. Journal of Biotechnology, 2013,168(2):149-154.
[41] Craig K, Johnson B R, Grunden A. Leveraging Pseudomonas Stress Response Mechanisms for Industrial Applications [J]. Frontiers in Microbiology, 2021,12:660134.
[42] Alfei S, Caviglia D, Piatti G, et al.activity of a self-biodegradable lysine-containing dendrimer against clinical isolates ofgenus [J]. International Journal of Molecular Sciences, 2021,22(14):7274.
[43] 肖 晶,郭丽英,赵 永,等.一株高效除磷的不动杆菌根瘤菌及其应用:中国, 201910185330.5 [P]. 2019-05-17.
Xiao J, Guo L Y, Zhao Y, et al. An efficient phosphorus removal strain ofand its application: China, 201910185330.5 [P]. 2019-05-17.
The stability of microbiological indicators in aerobic and anaerobic environment.
ZHANG Yang1, ZHENG Qian-xing1,2, WU Ren-ren1*, LI Guo-dong1, ZHONG Yi1**, CHENG Zhong-ying1, WEI Si-ye1, LI Kai-ming1
(1.South China Institute of Environmental Sciences, Ministry of Environmental Protection, Guangzhou 510655, China;2.Faculty of Environment Science and Engineering, Kunming University of Science and Technology, Kunming 650500, China)., 2022,42(3):1327~1334
We investigated the variations of pig-associated genetic markers, several microbiological indicators and potential pathogens in anaerobic and aerobic treatments using qPCR and Illumina sequencing methods. The six indicators (Streptococcaceae、Lactobacillaceae、Carnobacteriaceae、Ruminococcaceae、Lachnospiraceae and Prevotellaceae) had a similar decay trend in anaerobic and aerobic treatments, but these taxa persisted longer in anaerobic than aerobic treatments. Hence, the six indicators all can be used for identifying the recent pollution issues in aerobic water environment. The genetic markers including GenBac and Pig-2-Bac showed biphasic decay model in both aerobic and anaerobic treatments, and the first decay stage contributed the majority decay. However, the decay rates of genetic markers were significantly higher in aerobic than anaerobic treatments, which suggests the poor stability of the markers in aerobic water environment. For the identified potential pathogens, thegenetic markers presented higher correlation with,and(<0.05), but onlyshowed similar correlation coefficient in both anaerobic and aerobic treatments (0.913~0.953), which demonstrate the usefulness of thegenetic markers for predicting the presence of.
fecal pollution;microbial source tracking (MST);genetic markers;indicators;stability
X172
A
1000-6923(2022)03-1327-08
张 杨(1988-),男,宁夏银川人,助理研究员,博士,主要从事环境微生物研究.发表论文7篇.
2021-08-09
广东省自然科学基金资助项目(2020A1515011109);广州市科技计划项目(202002030377);中央级公益性科研院所基本科研业务专项(PM-zx703-202104-070,PM-zx703-202104-128,PM-zx703-202104-064)
*责任作者, 正高级工程师, wurenren@scies.org;**高级工程师, zhongyi@scies.org
猜你喜欢
节能与环保(2022年4期)2022-06-02
浙江大学学报(农业与生命科学版)(2021年6期)2022-01-08
科技视界(2021年21期)2021-08-24
渔业致富指南(2020年7期)2020-12-19
科学与信息化(2020年11期)2020-06-19
山东工业技术(2016年15期)2016-12-01
中外医疗(2016年29期)2016-11-30
中国实用医药(2016年1期)2016-01-11
湖南大学学报·自然科学版(2014年3期)2014-12-30
中国当代医药(2014年15期)2014-08-08
