
水中有机物对紫外光催化还原溴酸盐的影响
2022-03-29 10:44胡航恺徐浩丹卢晓辉王立章
中国环境科学 2022年3期
胡航恺,徐浩丹,卢晓辉,王立章,马 军,宋 爽,汪 达,*
水中有机物对紫外光催化还原溴酸盐的影响
胡航恺1,徐浩丹2,卢晓辉2,王立章3,马 军2,宋 爽1,汪 达1,3*
(1.浙江工业大学环境学院,浙江省工业污染微生物控制技术重点实验室,浙江 杭州 310032;2.哈尔滨工业大学环境学院,城市水资源与水环境国家重点实验室,黑龙江 哈尔滨 150006;3.中国矿业大学环境与测绘学院,江苏 徐州 221116)
通过一步水热法制备了TiO2与TiOF2用于考察不同水质条件下紫外光催化控制还原BrO3−的效率.结果表明,TiO2在纯水中还原BrO3−的效率(120min还原78.5%)显著高于TiOF2(120min还原57.0%).但当体系中存在难降解有机物(莠去津)或天然有机物(腐殖酸)时,TiOF2还原BrO3−的效率则优于TiO2.TiOF2尤其在20μmol/L莠去津和20μmol/L BrO3−共存条件下展现了良好的同步降解莠去津和还原BrO3−的特性,在反应120min后,莠去津的降解率和BrO3−的还原率分别达到了48.5%和99.0%.此外,TiOF2比TiO2表现出更强的抗水中有机物干扰的能力.当溶液中腐殖酸浓度为5mg/L时,TiO2和TiOF2体系中BrO3−的还原率分别为13.8%和29.8%,后者表现出更为稳定的BrO3−还原效率.在紫外光深度净水体系中,TiOF2具有更强抗水质干扰能力,更能适应水质条件变化带来的影响.
UV光催化;溴酸盐控制;TiOF2;难降解有机物;天然有机物
研究表明,多种难降解有机物可突破现有城市污水厂处理屏障,进入饮用水水源,对饮用水水质安全产生隐患[1−4].因此饮用水深度处理净化技术在提标改造工程中被大量使用[5].高级氧化技术作为饮用水提标改造技术之一,因其诸多优势而受到青睐[6].但水源水中含有一定量的溴离子(Br−),臭氧,过硫酸盐,芬顿等高级氧化技术均具有强氧化性,易使Br−转化成溴酸盐(BrO3−).天然水体中的Br−浓度一般为0.01~6.0mg/L[7],例如太湖水中的Br−浓度常年在120~180μg/L[8],采用臭氧氧化技术极易造成BrO3−超标.BrO3−被国际癌症研究中心(IARC)列为2B致癌物,并且我国《生活饮用水卫生标准》(GB5749-2006)要求饮用水中BrO3−浓度限值为10μg/L[9].UV光催化技术被证明能有效将BrO3−还原为Br−,从而降低水中BrO3−的浓度,是理想的饮用水BrO3−去除技术.在UV光的驱动下,光催化材料可被激发产生电子和空穴,材料表面的光生电子可将BrO3−还原为Br−,产生的空穴具有氧化性,可氧化水中的其他物质[10].由于UV光催化体系中同时存在氧化和还原途径,为了有效控制BrO3−,适宜的光催化剂的选取是该体系的核心.
目前最常见的用于UV光催化还原BrO3−研究的光催化剂为二氧化钛(TiO2),TiO2能在UV光照射下将BrO3−还原为Br−,但光照产生的空穴会生成羟基自由基,将Br−重新氧化生成BrO3−,因而效果有限[11];水中低浓度的天然有机物会影响BrO3−还原,例如0.5mg/L的腐殖酸(Humic Acid,HA)就可使Br−的生成量极大降低[12].但当BrO3−和布洛芬共存时,前者的还原率和后者的降解率均较其单独存在溶液中时有显著上升[13].因此推测水中不同种类和浓度有机物的存在会显著影响BrO3−的还原规律.目前UV光催化还原BrO3−的研究主要在纯水体系中开展,而对水中共存有机物的影响研究较少[14-15].
本文通过一步水热法制备了UV光催化材料钛氟氧化物(TiOF2)纳米方块,以锐钛矿TiO2作为参照材料,通过向体系中投加难降解有机物莠去津(ATZ)或易降解有机物HA,考察不同典型有机物对2种催化材料UV光催化还原BrO3−效率的影响,分析TiOF2纳米方块还原BrO3−的机理.
1 材料与方法
1.1 试剂
氢氟酸(³40.00%),溴化钾(优级纯),钛酸丁酯(TBOT,³99.00%),异丙醇(IPA,色谱级),草酸铵(AO,优级纯)和硝酸银(AgNO3,ACS纯)购自阿拉丁试剂有限公司;无水乙醇(分析纯)和冰乙酸(分析纯)购自天津天力化学试剂有限公司;溴酸钾(分析纯)购自国药集团化学试剂有限公司;莠去津(99%)购自美国Sigma-Aldrich公司.HA购自国际腐殖酸协会,第II类标准品,产品编号2S101H.气体(氮气,99.999%)由黎明气体公司提供.超纯水产自MillQ system.
1.2 催化剂制备方法
TiOF2纳米方块采用一步水热法反应合成.将25mL的TBOT与9.4mL的HF及50mL冰乙酸依次加入100mL聚四氟乙烯内衬的反应罐中混合均匀,再将反应罐密闭于不锈钢反应釜中,200℃进行12h的水热反应,反应结束后自然冷却到室温.得到的白色前驱体依次用蒸馏水以及乙醇溶液洗去杂质,所得的白色沉淀物在60℃烘箱中干燥过夜,所得的粉末即为TiOF2纳米方块.
锐钛矿TiO2纳米颗粒的合成方法与TiOF2纳米方块相同,但以相同体积的超纯水代替HF及冰乙酸.
1.3 材料表征方法
对反应制得的TiOF2和TiO2进行物理化学的测试表征.样品晶体微观结构采用X射线衍射分析(XRD,X-Pert,荷兰PANalytical公司)进行表征;样品的表面形貌结构采用场发射扫描电子显微镜(FE-SEM,S-4800,日本Hitachi公司)观察;样品的比表面积和孔径分布分别采用Brunauer-Emmett- Teller (BET)法和Barrett-Joyner-Halenda (BJH)法在物理吸附仪(ASAP 2020,美国Micromeritics公司)上测定.样品表面化学态和价带位置采用X射线光电子能谱(XPS,PHI 5000C,美国PerkinElmer公司)分析.样品的瞬态光电流测试在电化学工作站(CHI 660D,中国Chenhua公司)上完成,采用三电极体系,电解液为0.1mol/L Na2SO4,光源与反应实验光源相同.对电极和参比电极分别为Pt丝和Ag/AgCl电极.工作电极的制作方法如下:将10mg样品加入2mL超纯水和乙醇(1:1)溶液中,再加入50μL Nafion溶液(5wt%),超声混匀.取100μL悬浮液均匀涂抹在1cm2的FTO玻璃上,将电极放入60℃的真空烘箱中干燥6h.样品的光致发光(PL)光谱采用稳态荧光光谱仪(FLS920,英国Edinburgh公司)测定,激发波长250nm.
1.4 实验装置与方法
反应装置如图1所示,该装置由光源、光催化反应器和水冷系统3部分组成.光源为253.7nm 10W低压汞灯,光强为2.9×10−6Einstein/L s.反应器为圆柱形,实际有效容积约为550mL,侧壁上设取样口.反应器置于冷却罐中,冷却罐与低温恒温槽连通,配置冷凝水回流装置,以控制反应器温度为25℃.
反应前先向反应器中加入20μmol/L的BrO3−溶液,随后投入0.1g/L催化剂,插入事先预热30min的紫外灯开始反应.反应期间间隔适当时间用注射器取样,并用0.22μm滤膜滤去催化剂后分别测定溶液中的Br−,BrO3−和OBr−.Br−和BrO3−采用Dionex 3000离子色谱检测,通过标准曲线方法确定Br−和BrO3−的浓度,OBr−用N,N-二乙基对苯二胺/辣根过氧化物酶法(DPD/POD)测定[16],通过标准曲线方法确定OBr−的浓度.
考察ATZ或HA存在的影响时,将一定浓度的ATZ或HA与20μmol/L BrO3-溶液混合后一起加入反应器中启动反应.溶液中的ATZ剩余浓度采用高效液相色谱Waters 2695分析,通过标准曲线方法确定ATZ浓度.溶液中HA的剩余浓度通过紫外可见分光光度法测定,以UV254表示.
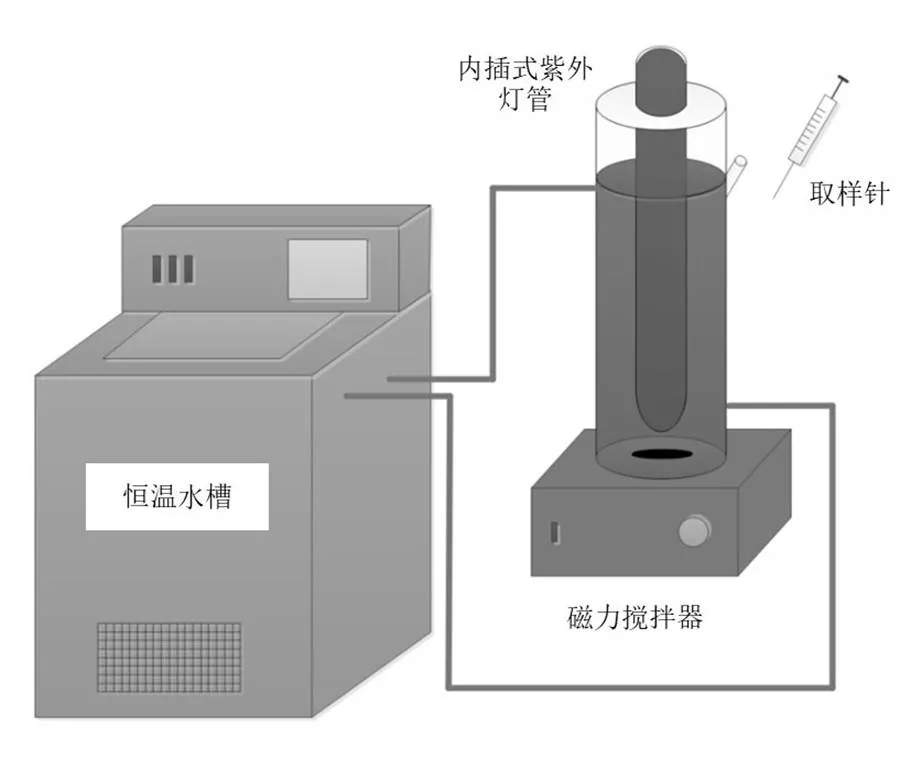
图1 紫外光催化反应装置
2 结果与讨论
2.1 材料表征
2.1.1 XRD分析 由图2可知,TiO2样品为锐钛矿相,与JCPDS(71-1167)标准卡一致,为四方晶系,空间群为I41/amd,空间群号141,没有发现属于金红石相或板钛矿相对应的峰.TiO2样品的主要暴露晶面为(101),(004)和(200)晶面,这与标准卡上的主要暴露晶面相吻合.TiOF2样品则与JCPDS(08-0060)标准卡一致,为立方体晶系,空间群为Pm-3m,空间群号221,无其他含氟化合物的杂峰,所有暴露晶面强度均与标准卡片接近,证明TiOF2样品成功合成[17].
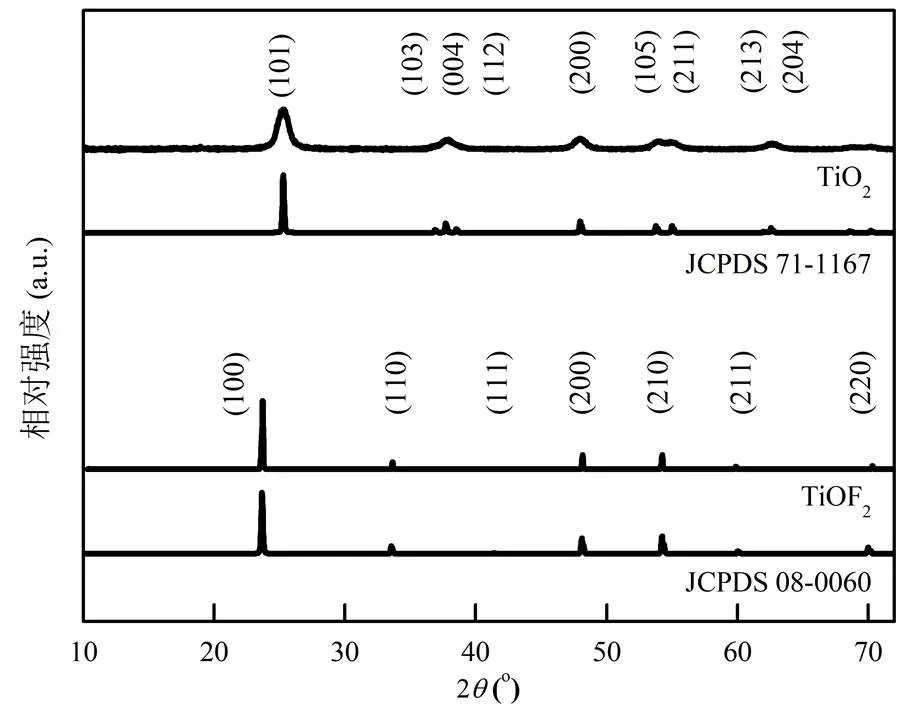
图2 TiO2和TiOF2样品的XRD及对应的标准卡片
2.1.2 形貌分析 如图3所示,TiO2的形貌为堆积的纳米颗粒,粒径为20~30nm,这是由于制备时未对其进行分散处理.TiOF2的形貌为正方形,表面光滑,边长为400~600nm.TiOF2较大的颗粒粒径会导致其比表面积的减小.

图3 TiO2和TiOF2样品的FE-SEM图
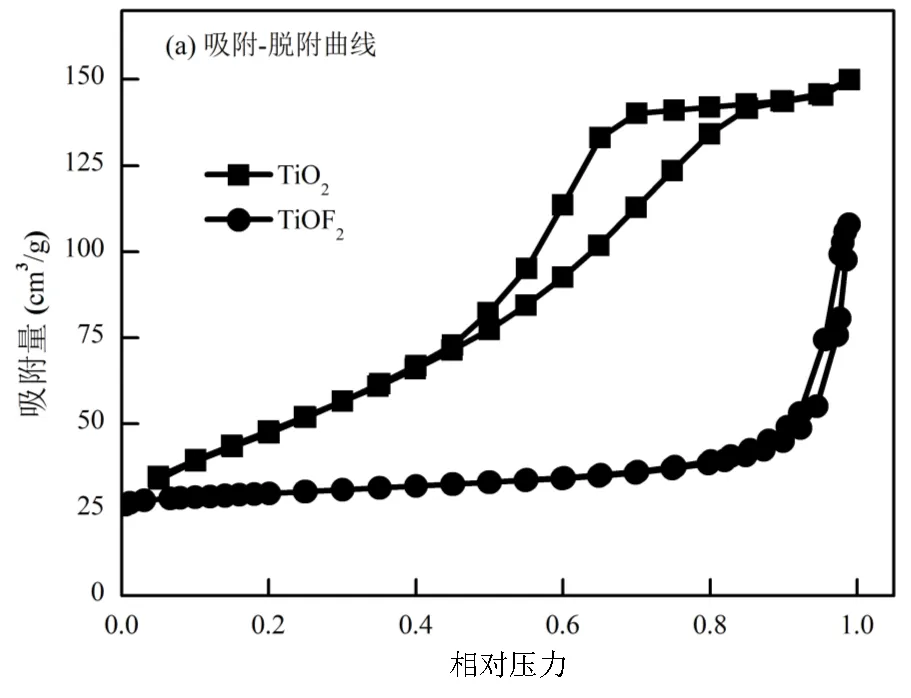
2.1.3 比表面积及孔径分析 TiO2和TiOF2的比表面积分别为177.5,8.1m2/g.TiOF2比表面积的减小与其较大的粒径有直接关系.如图4所示,TiO2和TiOF2的吸附等温线类型均为IV型,并在吸附中段出现滞后回环,说明在吸附过程中出现表面毛细凝聚现象[18].TiO2样品在相对吸附压力为0.85处闭合,并出现了一个平台,而TiOF2样品在吸附压力达到最大时仍未出现平台,而是直接作为滞后回环的终点,说明TiO2样品在毛细凝聚过程结束后,并未进一步形成多分子层的吸附,TiOF2样品则可视为仍未达到毛细凝聚饱和.
TiO2样品的滞后回环类型为H2型,TiOF2样品则为H3型,其中H2型为典型的墨水瓶型孔,而H3型为裂缝型孔或楔形孔,这说明钛氧化物的孔结构受氟化影响显著,从根本上改变了孔结构与孔形状[19].TiO2和TiOF2样品的脱附曲线开环位置分别位于相对压力0.45和0.87处,表明表面氟化可以使滞后回环向压力更高的区域移动,即说明孔径的分布也对应变大.
2.1.4 紫外可见漫反射(UV-vis DRS)分析 如图5所示,与TiO2相比,TiOF2在可见光区域(>420nm)有较为明显的吸收区间.但TiO2和TiOF2的吸收带边较为接近,对应的禁带宽度(Eg)分别为3.04,2.99eV,两者均需要由波长小于410nm的UV光激发.
TiO2和TiOF2导带(CB)位置的确定采用经验公式(1)计算,价带(VB)采用公式(2)计算.
CB=–C–g/2 (1)
VB=g+CB(2)
式中:为半导体内部各原子绝对电负性的几何平均值(Ti, O, F的电负性分别为3.45, 7.54, 10.41eV),C为常数(4.5eV)
计算得到TiO2和TiOF2的导带分别为−0.3, 1.27eV,价带分别为2.7,4.26eV,与之前的报道相符[20].
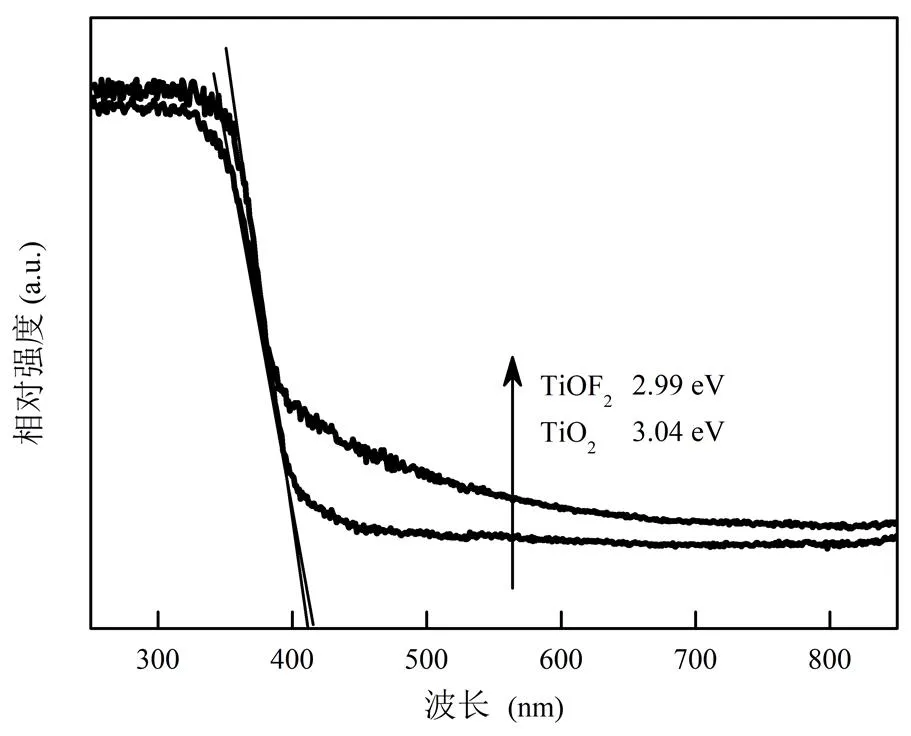
图5 TiO2和TiOF2样品的UV-vis DRS图
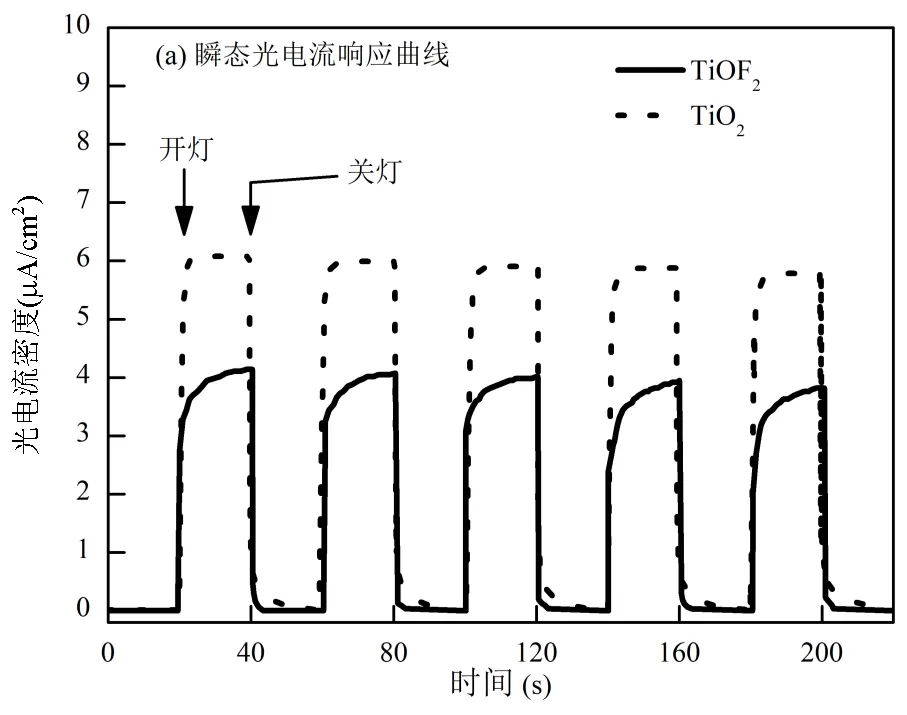
2.1.5 瞬态光电流响应测试分析 TiOF2和TiO2的瞬态光电流响应测试见图6(a),在每20s的开关灯循环下,TiO2和TiOF2均表现出明显的光电效应,证明2种材料均可被UV光激发产生光生电子并向外部迁移.TiO2比TiOF2表现出更高的光电流密度,说明在UV光激发下,TiO2的电荷转移速率更高.2种样品的PL谱如图6(b)所示,在260nm激发波长下,TiO2和TiOF2样品均在300~600nm之间存在连续的发射峰.TiO2和TiOF2样品的最大发射峰均在407nm左右(约等于3.0eV左右的带隙宽度),对应样品带隙的自由激发,这也与UV-vis DRS谱图的结果相吻合.图中的其他发射峰可能来自于激发产生的间接跃迁或材料内部的氧空穴和缺陷位点的激发[21].TiO2的PL谱强度整体略低于TiOF2样品,说明TiO2具有更低的电子-空穴复合速率和更快的光生电子分离传递速率[22].
2.2 催化结果分析
2.2.1 催化剂吸附性能测试 为排除吸附作用对催化剂还原BrO3−效率测试的影响,考察了2种催化剂对初始浓度为20μmol/L时的Br−和BrO3−的静态吸附效果,但吸附效果不明显(数据未列出).因此将Br−和BrO3−的初始浓度降低至5μmol/L进一步考察吸附效果,所得结果如图7.向含有5μmol/L的Br−中分别投加0.1g/L TiO2或TiOF2,在30min吸附过程中Br−的吸附量均小于4%,说明2种催化材料表面均不能有效吸附Br−;BrO3−不能大量吸附在催化材料表面,因此认为在UV光催化还原BrO3−过程中,TiO2和TiOF2对Br−和BrO3−的吸附作用较小,可被忽略.
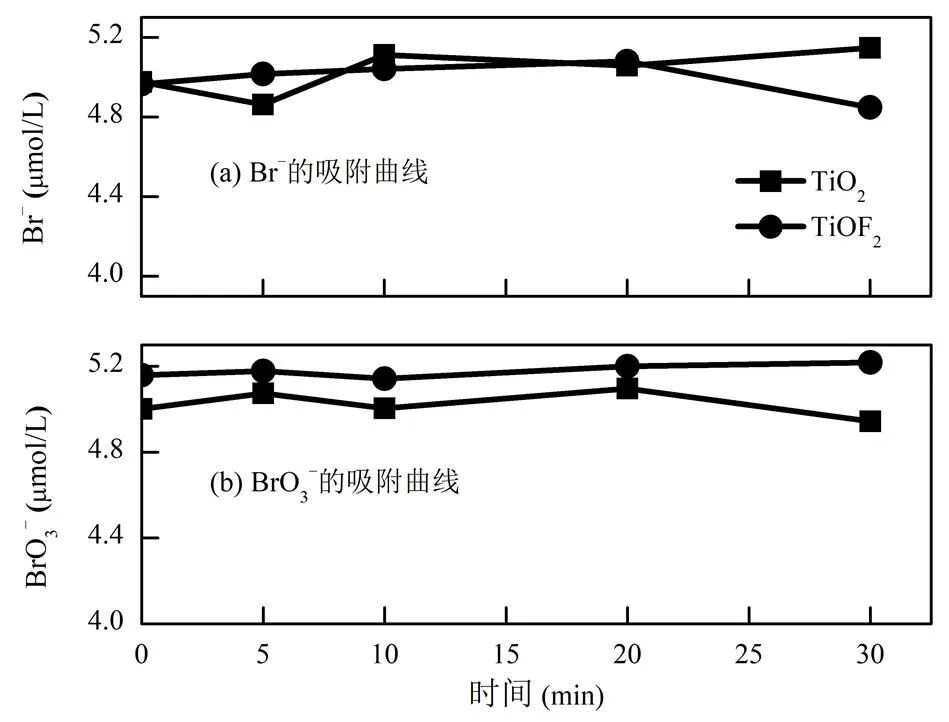
图7 TiO2和TiOF2吸附Br−和BrO3−的效果
[Br−]0= 5μmol/L, [BrO3−]0= 5μmol/L, [TiO2]0= 0.1g/L, [TiOF2]0= 0.1g/L,= 293K, pH = 6.8
2.2.2 纯水中催化剂还原BrO3−效率 由图8(a)可知,Br−的浓度在UV光体系中基本保持不变,且体系中也未检测到其他含溴物质,说明在UV光照Br−体系中Br−可以稳定存在,不会发生氧化还原反应.
直接UV光照还原20μmol/L BrO3−的效率如图8(b)所示,经过120min UV光照射后,BrO3−浓度降至11.4μmol/L,而Br−上升至8.0μmol/L,同时还生成了0.65μmol/L左右的HOBr/OBr−,说明单独UV光具有一定的BrO3−还原能力.向20μmol/L BrO3−溶液中投加TiO2后(图8(c)),BrO3−的还原效率与UV直接光照相比得到了显著提升,在反应结束时剩余的BrO3−浓度降至4.1μmol/L,对应Br−浓度则为15.9μmol/L,另有0.14μmol/L的HOBr/OBr−生成.在TiO2光催化剂的存在下,UV光可被TiO2吸收并激发生成光生电子-空穴对,其中的光生电子可由TiO2的导带导出并传递给固液界面附近的BrO3−,使得BrO3−被还原至Br−,而体系中少量存在的HOBr/OBr−是BrO3−还原的关键中间产物,证明了BrO3−被还原至Br−过程中的多电子转移过程(公式(3)~(5))[11].且通常认为BrO3−还原至HOBr/OBr−是BrO3−还原中的控速步骤,当HOBr/OBr−生成后,继续还原至Br−则较为容易.TiO2在纯水中展现出了优秀的UV光催化还原BrO3−能力,而相同投量TiOF2的BrO3−还原能力(图8(d))则不如TiO2,在120minUV光照后仍剩余8.6μmol/L BrO3−.TiO2和TiOF2的禁带宽度均为3.0eV左右,TiO2和TiOF2的导带位置分别为−0.3, 1.27eV,均高于BrO3−/Br−(1.42VNHE),BrO3−/ BrO−(1.47V)和BrO−/Br−(1.33V)的还原电位[23],且通常目标物在非均相表面还原前以吸附态存在,其还原电势会进一步降低[24-25].因此TiO2和TiOF2的导带上产生的电子均能有效还原BrO3−.TiO2和TiOF2的价带位置分别为2.7,4.26eV,与TiO2相比,TiOF2具有更正的价带位置,证明TiOF2价带上产生的空穴氧化能力比TiO2更强.而纯水BrO3−还原体系中,光催化材料展现的氧化性主要作用于Br−氧化及水氧化,若光催化剂展现的氧化性较强,则会加快Br−氧化重新生成BrO3−这一途径(公式(6)~(14)),从而导致BrO3−还原的效果被抑制,这也是TiOF2还原BrO3−效率弱于TiO2的主要原因[11].
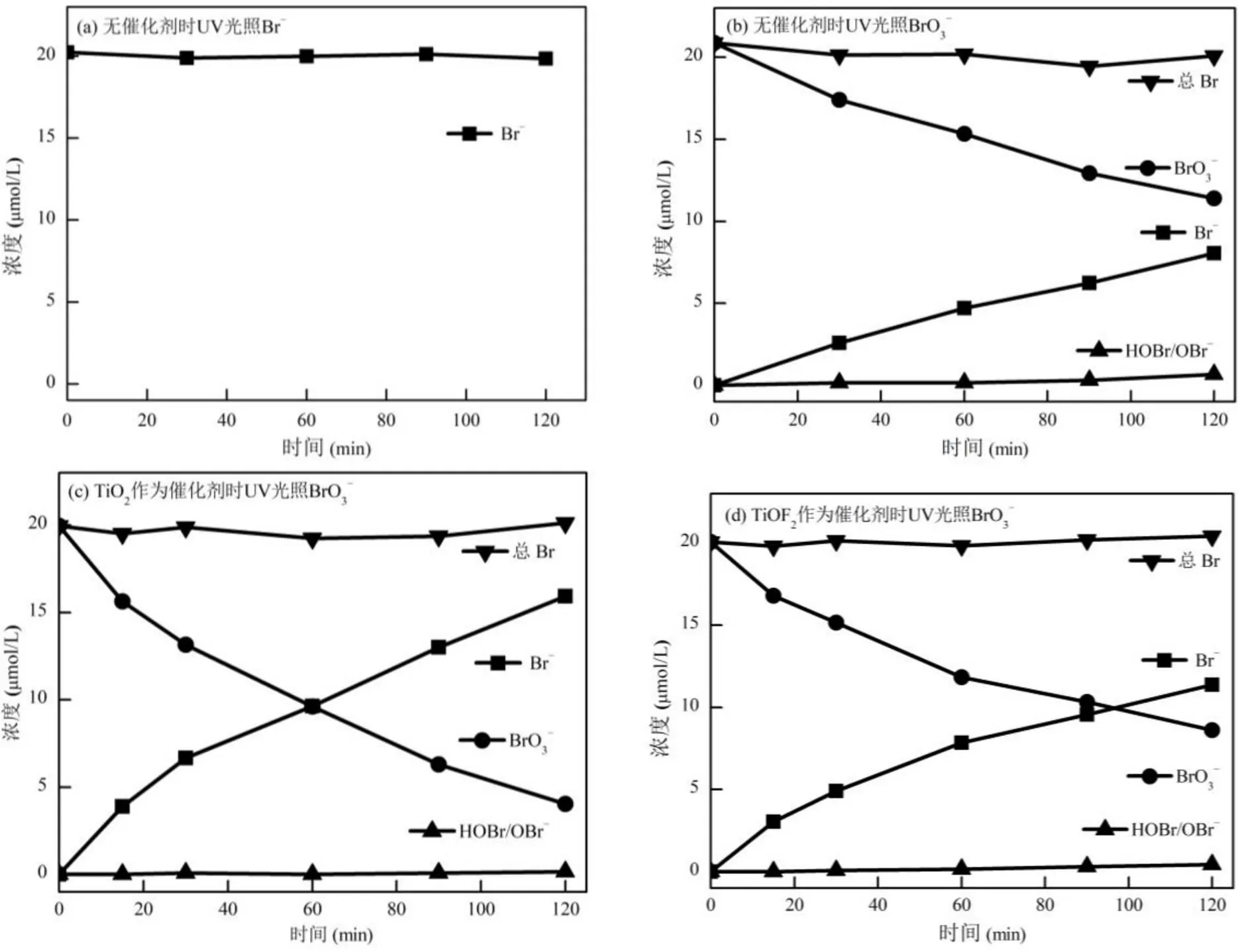
图8 UV光照下无催化剂、TiO2和TiOF2还原Br−或BrO3−时溶液中含溴物质浓度变化
[Br−]0= 20μmol/L, [BrO3−]0= 20μmol/L, [cat]0= 0.1g/L,= 293K, pH = 6.8
BrO3-+ e-+ H2O → BrO2-+ O2-(3)
BrO2-+ e-+ H2O →OBr-+ HO-(4)
OBr-+ e-+ H+ → Br-+ OH· (5)
Br-+ OH· →Br· + OH-(6)
Br-+ h+→Br· (7)
Br· + Br-→Br2·-(8)
2Br2·-→Br-+ Br3·-(9)
Br3·-+ H2O →HOBr + 2Br-+ H+(10)
OH· + HOBr →BrO· + H2O (11)
2BrO· + H2O →OBr-+ BrO2-+ 2H+(12)
BrO· + BrO2-→OBr-+ BrO2(13)
OH· + BrO2→BrO3-+ H+(14)
4组反应过程中的溴平衡核算发现,体系中的总溴浓度几乎保持不变,说明在反应过程中,Br−和BrO3−是主要的溴化物,而HOBr/OBr−是浓度较高的中间态溴化物,三者之间的转变共同稳定了溴平衡,Br2等其他溴化物未在体系检出.
采用牺牲剂法对TiOF2还原BrO3−过程体系中的可能活性物种进行了鉴定(图9).分别使用异丙醇(IPA)、草酸铵(AO)和AgNO3作为羟基自由基(∙OH),空穴(h+)和电子(e−)的牺牲剂.与无牺牲剂时相比,IPA和AO的加入明显加速了BrO3−的还原,表现为Br−生成量的增加,由120min反应后无牺牲剂时11.4μmol/L的Br−生成量提升至15.4,16.7μmol/L,说明体系中∙OH和h+的存在对BrO3−还原有明显的抑制作用.反之,e−牺牲剂AgNO3的引入则显著降低了Br−的生成量,说明光生电子是BrO3−还原的主要贡献者.
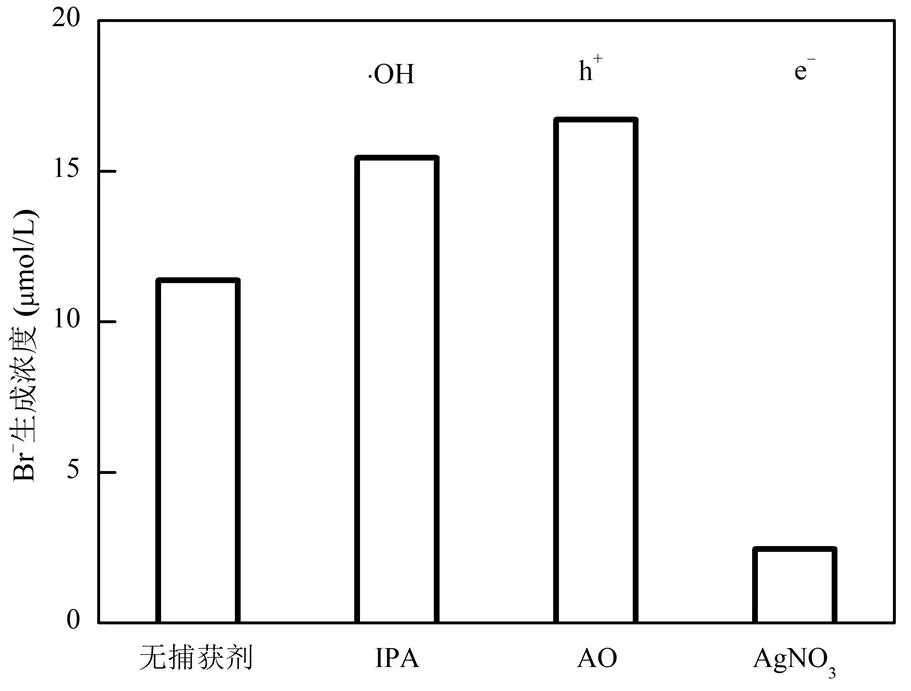
图9 不同抑制剂对UV光催化TiOF2还原BrO3−生成Br−的影响
[BrO3−]0= 20μmol/L, [cat]0= 0.1g/L,= 293K, pH = 6.8, [IPA]0= [AO]0= [AgNO3]0= 20μmol/L,= 120min
2.2.3 有机物存在时催化剂还原BrO3−效率 如图10(a)所示,在保持BrO3−初始投加浓度(20μmol/L)不变的条件下,分别向体系中添加1~20μmol/L的ATZ.UV直接光照组的Br−生成量随ATZ的投加量增加显著降低,从1μmol/L ATZ时的6.1μmol/L逐渐降至20μmol/L ATZ时的1.7μmol/L,原因为ATZ与BrO3−两者存在对UV光的竞争吸收,从而导致BrO3−直接接收到的光总量降低.而在TiO2和TiOF2体系中,Br−生成量均随ATZ的投加量增加而升高,且TiOF2体系中Br−生成量增加的速率高于TiO2体系,当ATZ投加量为20μmol/L时,TiO2和TiOF2体系中Br−生成量分别为17.7,19.8μmol/L.ATZ存在时Br−生成量的增加原因主要有:首先,与HA等富电子易降解有机物不同,ATZ等难降解有机物无法被UV光有效激发,亦不会与BrO3−竞争夺取体系中形成的电子[26];其次,ATZ存在时体系中空穴等氧化性物种氧化Br−重新生成BrO3−的反应途径被弱化.UV光催化过程中产生的空穴等氧化物种会与难降解有机物接触并使之降解,尤其是当难降解有机物的投量增加时,氧化物种与难降解有机物接触的几率增大,而与体系中已经还原生成的Br−的接触几率随之减小.此外,已知TiOF2产生的空穴氧化能力强于TiO2,因此在不同ATZ投量下TiOF2光催化降解ATZ的降解率均高于TiO2[27].
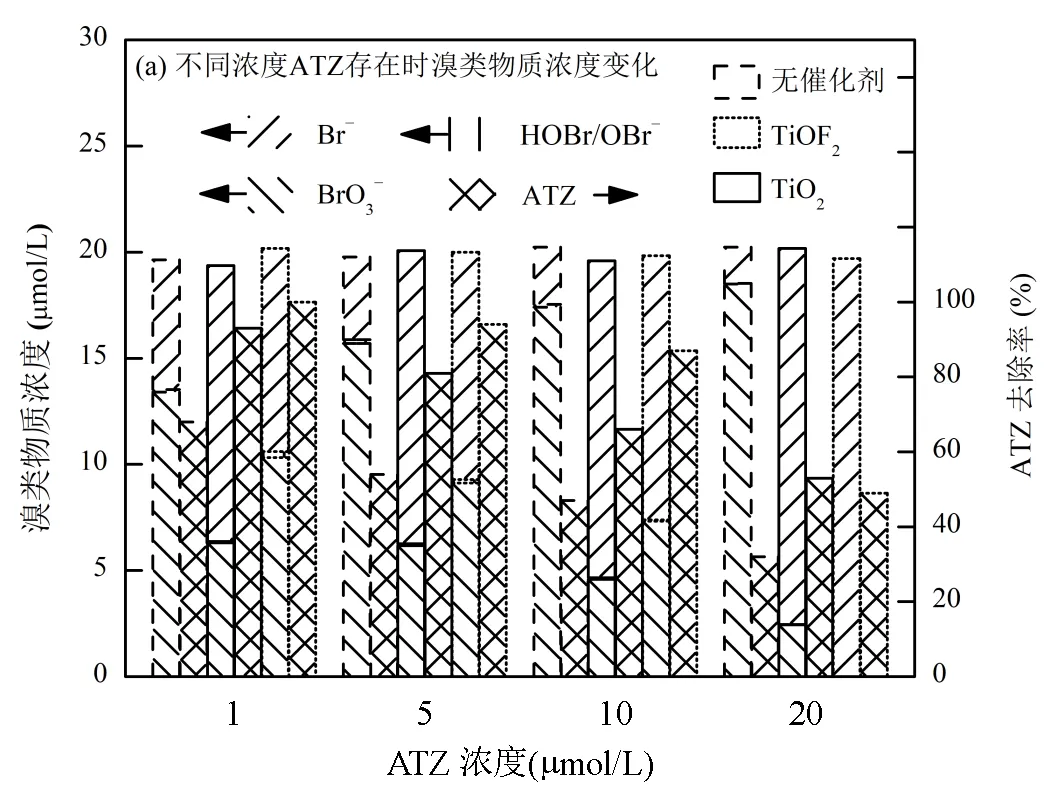
[BrO3−]0= 20μmol/L, [cat]0= 0.1g/L,= 293K, pH = 6.8,= 120min
与ATZ投加时的结果不同,向体系中投加不同浓度的HA后(图10(b)),所有反应体系中BrO3−的还原均受到抑制,且抑制程度随HA浓度的增加而显著增加,与相关研究结果相似[28].在直接UV光照条件下,Br−的生成量由无HA时的8.04μmol/L降至HA投量0.1mg/L时的6.97μmol/L,最后在HA投量5mg/L时仅剩0.74μmol/L.HA在光照条件下极为活泼,HA本身就能吸收光能转变为激发态或三重态,进一步消耗光催化产生的电子,进而抑制BrO3−还原过程[29];与此同时,光催化产生的空穴或其他氧化物种也能够从HA表面的富电子基团处夺得电子,进而使HA表面基团发生改变.因此HA的存在可同步消耗体系中的光生电子和空穴,并表现为BrO3−还原的抑制.此外,由于HA表面基团与结构成份复杂,因此采用UV254衡量HA的降解率,发现在所有实验中UV254的数值几乎保持不变,说明HA在该体系中并不会被大量降解.
与TiO2体系相比,HA存在时TiOF2UV催化还原BrO3−被抑制的程度较低,当HA投加量仅为0.5mg/L时,TiO2和TiOF2体系中Br−的生成量分别为9.68,11.27μmol/L,在HA投加量5mg/L时则为2.77,5.97μmol/L.在该体系的实验条件下,TiOF2还原BrO3−体系中对HA耐受性较高的原因可能是TiOF2的氟化表面(表面Ti-F键)降低了HA在TiOF2表面的吸附量,从而使得表面活性位被HA的覆盖度较低[30];相反,HA表面的-COOH等基团极易吸附在亲水性TiO2表面(表面Ti-OH基团),导致活性位被HA迅速覆盖,进而BrO3−无法与TiO2表面的活性位接触被还原.以上结论说明在水质条件较为复杂的情况下,UV光催化还原BrO3−体系中TiOF2比TiO2展现出更强的抗干扰能力.
2.2.4 催化剂稳定性研究 对同一批TiOF2进行重复性的BrO3−还原及ATZ降解实验,每次实验结束后将TiOF2以4000r/min离心分离后称重,质量不足的由新TiOF2补足再进行下一轮实验.由图11可知,在重复的5次实验过程中,BrO3−的还原量始终稳定在10.2~10.7μmol/L,对应ATZ的降解率保持在91%~ 95%.由于TiOF2表面并不强吸附Br−或BrO3−,且表面的F均以Ti-F的强化学键结合存在,因此TiOF2在UV光催化还原BrO3−过程中能够保持较高的稳定性.
2.2.5 催化机理解析 对不同水质条件下UV光催化还原BrO3−过程中TiOF2的作用机制进行解析(图12).TiOF2被UV光激发后,产生光生电子和空穴.在纯水体系中(图12(a)),由于体系中存在物种较为单一(只有溴化物),光生电子主要使BrO3−还原为Br−,而产生的光生空穴或其他氧化物种则会使生成的Br−或含溴中间产物重新被氧化生成BrO3−.而当BrO3−和难降解有机物(ATZ)共存于体系中时(图12(b)),由于难降解有机物难以和BrO3−共同竞争获得光生电子,因此BrO3−还原为Br−的效率基本保持不变,而一部分光生空穴则能与ATZ反应使之降解,同时降低了Br−与体系中氧化性物种的碰撞几率.该体系最终总体表现为Br−生成量的增加以及难降解有机物的有效降解.但若性质较为活泼的天然有机物(HA)与BrO3−共存时(图12(c)),HA将与溴化物同时争夺体系中生成的光生电子与空穴.HA表面的-COOH,-CHO等基团易夺得光生电子,而-NH2, -OH等基团则容易在空穴处被氧化,此外,HA本身具有较强的吸光性,降低了TiOF2接收到的光子数,以上因素综合显著抑制了BrO3−的还原速率[31].
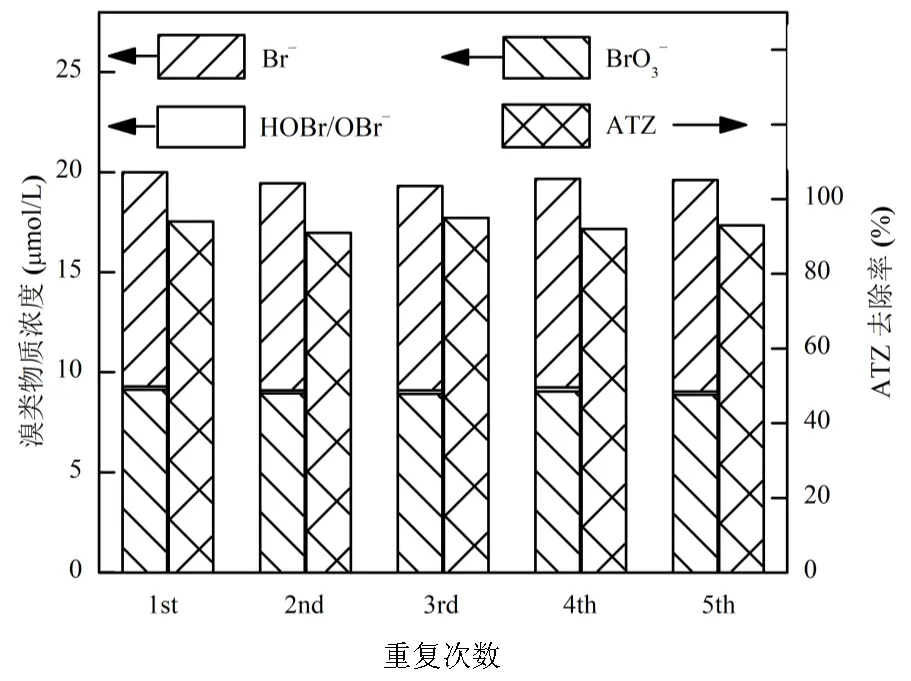
图11 难降解有机物存在时TiOF2对还原BrO3−的影响重复性实验
[BrO3−]0= 20μmol/L, [TiO2]0= 0.1g/L, [TiOF2]0= 0.1g/L, [ATZ]0= 5μmol/L,= 293K, pH = 6.8
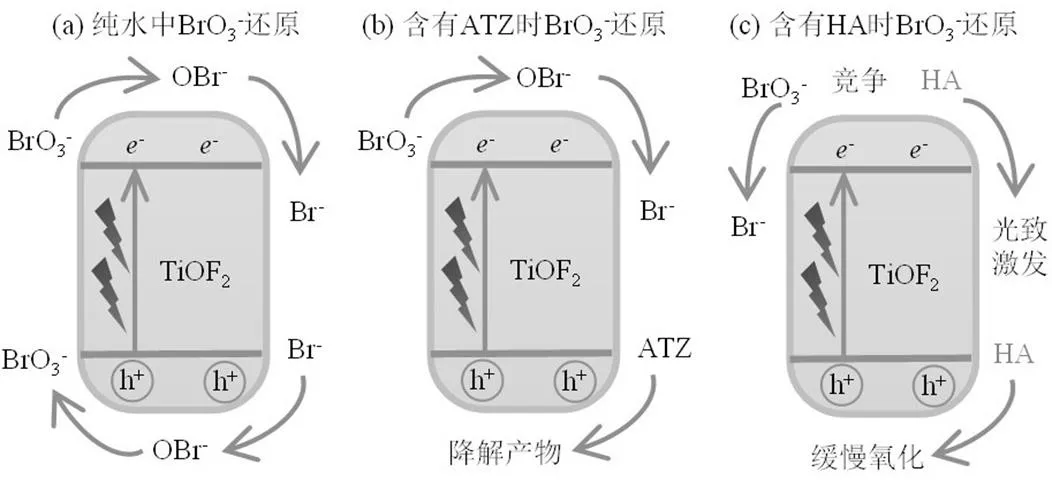
图12 TiOF2UV光催化还原BrO3−的反应机理
3 结论
3.1 TiO2形貌为堆积的纳米颗粒,粒径为20~ 30nm,比表面积为177.5m2/g,TiOF2的形貌为正方形,表面光滑,边长为400~600nm,比表面积为8.1m2/g.
3.2 纯水体系中,TiO2与UV光催化体系还原BrO3−的效率优于TiOF2.经过120min UV光照射后,TiO2将20μmol/L BrO3−浓度降至4.1μmol/L,生成15.9μmol/L的Br−和0.14μmol/L的HOBr/OBr−.相同条件下TiOF2仅使BrO3−浓度降至8.6μmol/L,生成11.2μmol/L的Br−.TiO2和TiOF2的价带位置分别为2.7,4.26eV,与TiO2相比,TiOF2具有更正的价带位置,证明TiOF2价带上产生的空穴氧化能力比TiO2更强,故TiOF2在纯水中的BrO3−还原能力次于TiO2.
3.3 在难降解有机物ATZ和BrO3−共存体系中,当ATZ和BrO3−投加量均为20μmol/L时,TiO2和TiOF2体系中Br−生成量分别为17.7,19.8μmol/L.ATZ消耗了部分光生空穴,从而降低了Br−被重新氧化生成BrO3−的几率.当HA与BrO3−共存时,尽管TiO2与TiOF2的还原BrO3−能力均受抑制,但TiOF2因表面具有的Ti-F键而表现出更强的抗水质干扰能力.
[1] Xie H, Hao H, Xu N, et al. Pharmaceuticals and personal care products in water, sediments, aquatic organisms, and fish feeds in the Pearl River Delta: Occurrence, distribution, potential sources, and health risk assessment [J]. Science of the total Environment, 2019,659:230-239.
[2] Liang M, Xian Y, Wang B, et al. High throughput analysis of 21perfluorinated compounds in drinking water, tap water, river water and plant effluent from southern China by supramolecular solvents- based microextraction coupled with HPLC-Orbitrap HRMS [J]. Environmental Pollution, 2020,263:114389.
[3] Jian Y, Yunting X, Xianghong T, et al. Atrazine and its metabolites (ATZs) in source water, finished water, and tap water from drinking water treatment plants and its human risk assessment in Zhoukou City, China [J]. Human and Ecological Risk Assessment: An International Journal, 2021,27(7):1-13.
[4] Di Carro M, Lluveras Tenorio A, Benedetti B, et al. An innovative sampling approach combined with liquid chromatography–tandem mass spectrometry for the analysis of emerging pollutants in drinking water [J]. Journal of Mass Spectrometry, 2020,55(11):e4608.
[5] 王 昊.UV/H2O2高级氧化法深度去除水中臭味物质 [J]. 中国给水排水, 2018,34(19):48-51.
Wang H. Deep removal of odor substances in water by UV/H2O2advanced oxidation [J]. China Water & Wastewater, 2018,34(19):48- 51.
[6] Applications of advanced oxidation processes (AOPs) in drinking water treatment [M]. Netherlands: Springer International Publishing, 2019:153-155.
[7] 林英姿,刘 栋.原水臭氧氧化生成溴酸盐的影响因素研究进展[J]. 中国资源综合利用, 2017,35(1):43-45.
Lin Y Z, Liu D. Study on the factors effecting the formation of bromate in ozone oxidation water [J]. China Resources Comprehensive Utilization, 2017,35(1):43-45.
[8] 杨蕴秀,李 攀,于水利,等.东太湖原水臭氧氧化过程中溴酸盐及溴代有机消毒副产物的控制[J]. 净水技术, 2017,36(6):16-21,29.
Yang Y X, Li P, Yu S L, et al. Control of bromate and bromo-organic disinfection by-products (DBPs) in ozone oxidation process for raw water of east Taihu lake [J]. Water Purification Technology, 2017,36 (6):16-21,29.
[9] GB 5749-2006 生活饮用水卫生标准 [S].
GB 5749-2006 Standards for drinking water quality [S].
[10] 朱欢欢,孙韶华,冯桂学,等.紫外联用高级氧化技术处理饮用水应用进展 [J]. 水处理技术, 2019,45(3):1-7,13.
Zhu H H, Sun S H, Feng G X, et al. Application progress of ultraviolet combined advanced oxidation technology in drinking water treatment [J]. Technology of Water Treatment, 2019,45(3):1-7,13.
[11] Zhang X, Zhang T, NgJ, et al. Transformation of bromine species in TiO2photocatalytic system [J]. Environmental Science & Technology, 2010,44(1):439-444.
[12] Zhang Y, Li J, Li L, et al. Influence of parameters on the photocatalytic bromate removal by F-graphene-TiO2[J]. Environmental Technology, 2019,42(2):248-256.
[13] Zhang Y, Li J, Liu H. Synergistic removal of bromate and ibuprofen by graphene oxide and TiO2heterostructure doped with F: Performance and mechanism [J]. Journal of Nanomaterials, 2020,2020:6094984.
[14] 黄 鑫,高乃云,卢 宁.BrO3−等溴类物质在长江水氯化过程中的形成 [J]. 中国环境科学, 2007,27(6):806-810.
Huang X, Gao N Y, Lu N. Formation of BrO3−et al. bromine kind matter in chlorination water of Yangtze River [J]. China Environmental Science, 2007,27(6):806-810.
[15] 周 娟,陈 欢,李晓璐,等.Pd/CeO2催化水中溴酸盐的加氢还原研究 [J]. 中国环境科学, 2011,31(8):1274-1279.
Zhou J, Chen H, Li X L, et al. Study on liquid phase catalytic hydrogenation of bromate over Pd/CeO2catalyst. [J]. China Environmental Science, 2011,31(8):1274-1279.
[16] Zhang Y, Li L, Liu H, et al. Graphene oxide and F co-doped TiO2with (001) facets for the photocatalytic reduction of bromate: Synthesis, characterization and reactivity [J]. Chemical Engineering Journal, 2017,307:860-867.
[17] Bader H, Sturzenegger V, Hoigné J. Photometric method for the determination of low concentrations of hydrogen peroxide by the peroxidase catalyzed oxidation of N, N-diethyl-p-phenylenediamine (DPD) [J]. Water Research, 1988,22(9):1109-1115.
[18] Wen C Z, Hu Q H, Guo Y N, et al. From titanium oxydifluoride (TiOF2) to titania (TiO2): phase transition and non-metal doping with enhanced photocatalytic hydrogen (H2) evolution properties [J] Chemical Communications, 2011,47(21):6138-6140.
[19] Cui X, Wang J, Zhang X, et al. Preparation of nano-TiO2by a surfactant-free microemulsion-hydrothermal method and its photocatalytic activity [J]. Langmuir, 2019,35(28):9255-9263.
[20] Miranda-Garcia N, Suarez S, Sanchez B, et al. Photocatalytic degradation of emerging contaminants in municipal wastewater treatment plant eluents using immobilized TiO2in a solar pilot plant [J]. Applied Catalysis B: Environmental, 2011,103(3/4):294-301.
[21] Dong P, Cui E, Hou G, et al. Synthesis and photocatalytic activity of Ag3PO4/TiOF2composites with enhanced stability [J]. Materials Letters, 2015,143:20-23.
[22] Cruz M R A, Sanchez-Martinez D, Torres-Martínez L M. TiO2nanorods grown by hydrothermal method and their photocatalytic activity for hydrogen production [J]. Materials Letters, 2019,237: 310-313.
[23] Liu Y, Ma Z. TiOF2/g-C3N4composite for visible-light driven photocatalysis [J]. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 2021,618:126471.
[24] You Y, Yuan H, Wu Y, et al. A novel red phosphorus/perylene diimide metal-free photocatalyst with p-n heterojunctions for efficient photoreduction of bromate under visible light [J]. Separation and Purification Technology, 2021,264:118456.
[25] Zhang Y, Xia B, Ran J, et al. Atomic-level reactive sites for semiconductor-based photocatalytic CO2reduction [J]. Advanced Energy Materials, 2020,10(9):1903879.
[26] Eftekhari A, Mohamedi M. Tailoring pseudocapacitive materials from a mechanistic perspective [J]. Materials Today Energy, 2017,6:211-229.
[27] Gondal M A, Suliman M A, Dastageer M A, et al. Visible light photocatalytic degradation of herbicide (Atrazine) using surface plasmon resonance induced in mesoporous Ag-WO3/SBA- 15composite [J]. Jourmal of Molecular Catalysis A: Chemical, 2016, 425:208-216.
[28] Zhang J, Tian P, Tang T, et al. Excellent photoelectrochemical hydrogen evolution performance of FeSe2nanorod/ZnSe 0D/1D heterostructure as efficiency carriers migrate channel [J]. International Journal of Hydrogen Energy, 2020,45(15):8526-8539.
[29] Santos S G S, Paulista L O, Silva T F C V, et al. Intensifying heterogeneous TiO2photocatalysis for bromate reduction using the NETmix photoreactor [J]. Science of the Total Environment, 2019, 664:805-816.
[30] Parker K M, Pignatello J J, Mitch W A. Influence of ionic strength on triplet-state natural organic matter loss by energy transfer and electron transfer pathways [J]. Environmental Science & Technology, 2013, 47(19):10987-10994.
[31] Mirzaei A, Chen Z, Haghighat F, et al. Enhanced adsorption of anionic dyes by surface fluorination of zinc oxide: A straightforward method for numerical soling of the ideal adsorbed solution theory (IAST) [J]. Chemical Engineering Journal, 2017,330:407-418.
[32] Xia Y, Liu X, Feng X. Asymmetric catalytic reactions of donor- acceptor cyclopropanes [J]. Angewandte Chemie, 2021,133(17):9276- 9288.
Influencing of aquatic organic matters on UV photocatalytic reduction efficiency of bromate.
HU Hang-kai1, XU Hao-dan2, LU Xiao-hui2, WANG Li-zhang3, MA Jun2, SONG Shuang1, WANG Da1,3*
(1.Key Laboratory of Microbial Technology for Industrial Pollution Control of Zhejiang Province, College of Environment, Zhejiang University of Technology, Hangzhou 310032, China;2.State Key Laboratory of Urban Water Resource and Environment, School of Environment, Harbin Institute of Technology, Harbin 150090, China;3.School of Environment Science and Spatial Informatics, China University of Mining and Technology, Xuzhou 221116, China)., 2022,42(3):1164~1172
TiO2and TiOF2were prepared by a one-step hydrothermal method and their efficiencies of BrO3−reduction under UV photocatalysis were investigated in water matrix. In pure water, 78.5% of BrO3−was reduced by UV/TiO2after 120min, which was significantly higher than that on UV/TiOF2(57.0%). While in water contained refractory organic pollutant (atrazine, ATZ) or natural organic matter (humic acid, HA), the results were opposite to those in pure water. UV/TiOF2achieved superior simultaneous ATZ degradation and BrO3−reduction when refractory organic pollutant co-existed with BrO3−. The ATZ degradation and BrO3−reduction rates reached 48.5% and 99.0% after 120min when 20 μmol/L ATZ and 20μmol/L BrO3−co-existed in the solution. The BrO3−reduction efficiencies of UV/TiO2and UV/TiOF2were 13.8% and 29.8% when 5mg/L of HA was added into the solution, demonstrating that compared with UV/TiO2, UV/TiOF2revealed robust BrO3−reduction ability in the complex water constituents. TiOF2presented stronger resistance to water matrix applied in UV photocatalytic advanced water purification system.
UV photocatalysis;bromate control;TiOF2;refractory organic pollutant;natural organic matter
X703.5
A
1000-6923(2022)03-1164-09
胡航恺(2000-),男,浙江温州人,浙江工业大学本科生,主要从事水污染控制研究.
2021-08-05
国家自然科学基金资助项目(22076168,52000158);浙江省自然科学基金青年基金资助项目(LQ21E080011);第69批中国博士后科学基金面上资助项目(2021M693414)
*责任作者, 副教授, wangda@zjut.edu.cn
猜你喜欢
科学之友(2022年11期)2022-11-03
力学学报(2022年5期)2022-06-16
辽宁石油化工大学学报(2021年6期)2022-01-04
车用发动机(2021年5期)2021-10-31
粉末冶金技术(2021年1期)2021-03-29
无机盐工业(2020年1期)2020-12-31
物理化学学报(2019年2期)2019-12-24
物理化学学报(2019年8期)2019-09-03
中国科技纵横(2019年3期)2019-03-25
分析化学(2017年12期)2017-12-25
