
正交试验法优化决明子的炮制工艺*
2022-03-28 01:31:42
右江民族医学院学报 2022年1期
(皖南医学院药学院,安徽 芜湖 241002)
决明子为豆科植物钝叶决明(CassiaobtusifoliaL.)或决明(小决明)(CassiatoraL.)的干燥成熟种子,具清热明目、润肠通便之功效,用于目赤涩痛、大便秘结等[1]。决明子始载于《神农本草经》,为上品,全国各省份多有栽培,分布广泛[2-3]。现代药理学研究证明,决明子除含有糖、蛋白质及多种微量元素如铁、锌等外,还富含大黄酚、大黄素等多种蒽醌类化合物[4]。现有研究表明,决明子蒽醌类成分具有护肝、调节免疫力、泻下等药理作用[5-6]。《中华人民共和国药典》2020年版(以下简称《中国药典》)记载了决明子的炮制工艺:取净决明子,照清炒法(通则0213),炒至微鼓程度,有香气[1]。这种炮制方法过程简单,操作方便,但因《中国药典》没有提供炮制工艺参数如炒制温度、炒制时间等可供参考,炒制结果凭经验掌握,炒制程度较难掌握,常出现“太过”或“不及”的现象[7],因而在决明子的实际炒制过程中,炒制程度取决于个人经验来判断,导致决明子的炒制质量参差不齐,进而影响其药理作用。本研究采用差示分光光度法建立决明子总蒽醌(含游离蒽醌)含量测定的方法,并采用正交试验法优化决明子的炮制工艺,确定决明子炮制工艺参数,以期进一步提升决明子的炮制质量。
1 材料与方法
1.1材料 决明子清炒品(批号:2019073101、2020071901、2019052702、2020052301、2019073103,北京同仁堂诚安药材有限公司);决明子(批号:20170310,河北安国市御颜坊中药材有限公司);丹蒽醌(批号:20200924,纯度≥97%,西亚化学科技有限公司);乙酸镁(批号:D4703,西亚化学科技有限公司);其他试剂均为分析纯。
1.2仪器 U-5100型分光光度计(日本株式会社高新技术科学那珂事业所);UV-5100紫外可见分光光度计(上海元析仪器有限公司);FA2004B电子天平(上海越平科学仪器有限公司);Heidolph-LR4010/4011旋转蒸发仪(德国海道尔夫公司);DRT-BRA型表显电热套(郑州长城科工贸有限公司);KQ5200E型超声波清洗机(昆山市超声仪器有限公司);AR590F非接触式红外测温仪(香港希玛仪表集团有限公司);GKC数显控温水浴锅(上海浦东电理仪器厂);RHP-400A型高速多功能粉碎机(浙江永康市荣浩工贸有限公司);KNT-II-20分析型超纯水机(合肥科宁特水处理设备有限公司);101-1-5电热恒温鼓风干燥箱(上海跃进医疗器械厂)。
1.3方法
1.3.1决明子样品溶液的制备(总蒽醌含量测定用) 取决明子清炒品适量粉碎,过50目筛。取决明子粉末40 g,精密称定,置圆底烧瓶中,按料液比(1∶12,g/ml)加入60%乙醇,回流提取3次,每次2 h。合并提取液并回收溶剂,提取物转移至蒸发皿中, 60 ℃烘干至恒重,得干膏,称重备用。取干膏2 g,精密称定,置圆底烧瓶中,加稀盐酸100 ml,水浴水解1 h,立即冷却,滤过,用二氯甲烷萃取四次,每次30 ml,合并二氯甲烷液,回收二氯甲烷,残渣用无水乙醇溶解,转移至50 ml量瓶中,以无水乙醇定容至刻度,摇匀,备用。
1.3.2丹蒽醌对照品溶液的制备 取丹蒽醌对照品10 mg,精密称定,用无水乙醇定容至50 ml量瓶中,摇匀备用。
1.3.30.5%醋酸镁-乙醇溶液的制备 取乙酸镁1.25 g,精密称定,用无水乙醇定容到250 ml量瓶中,振摇,充分溶解,备用。
1.3.4最大吸收波长的测定 精密吸取丹蒽醌对照品溶液和决明子样品溶液各2 ml于10 ml量瓶中,加0.5%醋酸镁-乙醇溶液,定容至刻度,摇匀。以等量无水乙醇加0.5%醋酸镁-乙醇溶液同法处理作参比;另取丹蒽醌对照品溶液和决明子样品溶液各2 ml于10 ml量瓶中,加无水乙醇,定容至刻度,摇匀。以无水乙醇作参比。分别于400~800 nm范围内进行光谱扫描确定最大吸收波长。
1.3.5标准曲线的绘制 精密吸取“1.3.2”项下丹蒽醌对照品溶液0.05 ml、0.1 ml、0.2 ml、0.4 ml、0.8 ml、1.6 ml于10 ml量瓶中,加0.5%醋酸镁-乙醇溶液,定容至刻度,摇匀。以无水乙醇加0.5%醋酸镁-乙醇溶液同法处理作参比。在最大吸收波长处测定吸光度。另取上述系列丹蒽醌对照品溶液,加无水乙醇,定容至刻度,摇匀。以无水乙醇作参比。在最大吸收波长处测定吸光度。计算丹蒽醌显色前后的吸光度差△A,以吸光度差△A为纵坐标,以浓度c为横坐标,绘制标准曲线。
1.3.6方法学考察
1.3.6.1精密度 精密吸取“1.3.1”项下样品溶液2 ml于10 ml量瓶中,按照“1.3.4”项下方法处理。连续测定5次,计算吸光度差△A及其RSD值。
1.3.6.2稳定性 精密吸取“1.3.1”项下样品溶液2 ml于10 ml量瓶中,按照“1.3.4”项下方法处理。在0 h、0.5 h、1 h、1.5 h、2 h处,测定吸光度,计算吸光度差△A及其RSD值。
1.3.6.3重复性 取同一批决明子清炒品粉末5份,按照“1.3.1”项下方法,制备成样品溶液,分别精密吸取样品溶液2 ml于10 ml量瓶中,按照“1.3.4”项下方法处理,测定吸光度,计算吸光度差△A及其RSD值。
1.3.6.4加样回收率 精密吸取已知浓度的决明子样品溶液2 ml,置于10 ml量瓶中,加入一定量丹蒽醌对照品溶液(丹蒽醌的质量为所加入样品溶液中总蒽醌质量的80%、100%和120%),共9组。按照“1.3.4”项下方法处理,测定吸光度,计算回收率及其RSD值。
1.3.6.5样品含量测定 取决明子清炒品5批,按照“1.3.1”项下方法,制备样品溶液,分别精密吸取样品溶液2 ml于10 ml量瓶中,按照“1.3.4”项下方法处理,测定吸光度,计算决明子总蒽醌含量。
1.3.7决明子炮制工艺的考察 取决明子生品适量,60 ℃干燥24 h。取100 g决明子,放入炒制容器内,不断翻炒,待药温达到规定温度范围开始计时,期间用红外测温枪测温,避免超出火候温度范围。将决明子炒制品粉碎过50目筛,取粉末5 g,精密称定,按“1.3.1”项下方法制备干膏并计算干膏得率(%),并按照“1.3.1”项下方法制备样品溶液,按“1.3.4”项下方法处理,测定,计算决明子总蒽醌的百分含量。
1.3.7.1游离蒽醌含量测定 经考察,“1.3.5”、“1.3.6”项下的标准曲线及其线性范围和方法学验证也适用于游离蒽醌含量测定。取“1.3.7”项下干膏0.5 g,置10 ml离心管内,加入2 ml纯化水,超声30 min。按照“1.3.1”方法萃取,回收二氯甲烷,定容至25 ml量瓶中,摇匀,按“1.3.4”项下方法处理,测定,计算决明子游离蒽醌的百分含量。
1.3.7.2正交试验因素水平表 以炒制方法(A)、炒制火候(B)和炒制时间(C)为因素,每个因素3个水平,因此选用L9(34)表安排正交试验[8]。因素水平见表1。

表1 因素水平
1.3.7.3蛤炒工艺的验证 对蛤粉炒工艺进行验证,平行炒制3份,并以《中国药典》(2020年版)的炮制工艺(清炒)作对比。
1.3.8数据处理
1.3.8.1评价指标 综合评价为游离蒽醌含量和总蒽醌含量的比值。
1.4统计学方法 采用SPSS 23.0统计软件对实验数据进行分析,对验证实验数据进行正态检验和两独立样本t检验分析,P<0.05为差异具有统计学意义。
2 结果
2.1总蒽醌(含游离蒽醌)含量测定方法的建立
2.1.1测定波长的选择 按照“1.3.4”项下方法将决明子样品和对照品分别于400~800 nm范围内进行光谱扫描确定最大吸收波长。结果显示对照品在514 nm处有最大吸收,决明子样品在514 nm处也有吸收,因此选择514 nm为测定波长。见图1。
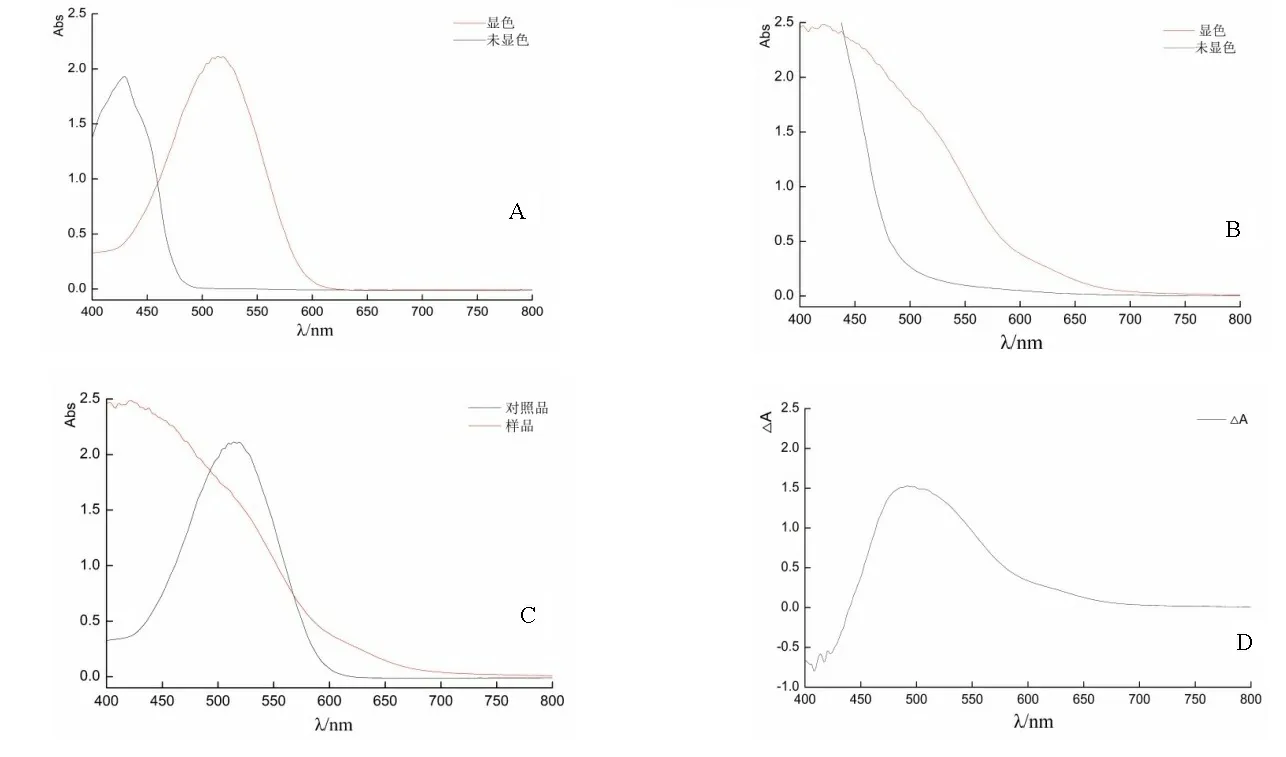
A:丹蒽醌对照品显色前后光谱图;B:样品显色前后光谱图;
2.1.2标准曲线的绘制 按照“1.3.5”项下方法,在514 nm处测定吸光度,计算丹蒽醌显色前后的吸光度差△A。以吸光度差△A为纵坐标,以浓度c(μg/ml)为横坐标,绘制标准曲线,得回归方程△A=0.0501c-0.0015,r=0.9999,线性范围1.04~16.64 μg/ml。结果表明吸光度之差△A与对照品浓度c线性关系良好。
2.1.3方法学考察
2.1.3.1精密度 按照“1.3.6.1”项下方法,得吸光度差△A,计算其RSD值为0.25%,结果表明该方法精密度良好。见表2。

表2 精密度结果
2.1.3.2稳定性 按照“1.3.6.2”项下的方法,得吸光度差△A,计算其RSD值为1.90%,结果表明样品溶液显色后2 h内较稳定。见表3。

表3 稳定性结果
2.1.3.3重复性 按照“1.3.6.3”项下的方法,得吸光度差△A,计算其RSD值为2.52%,结果表明该方法重复性良好,见表4。
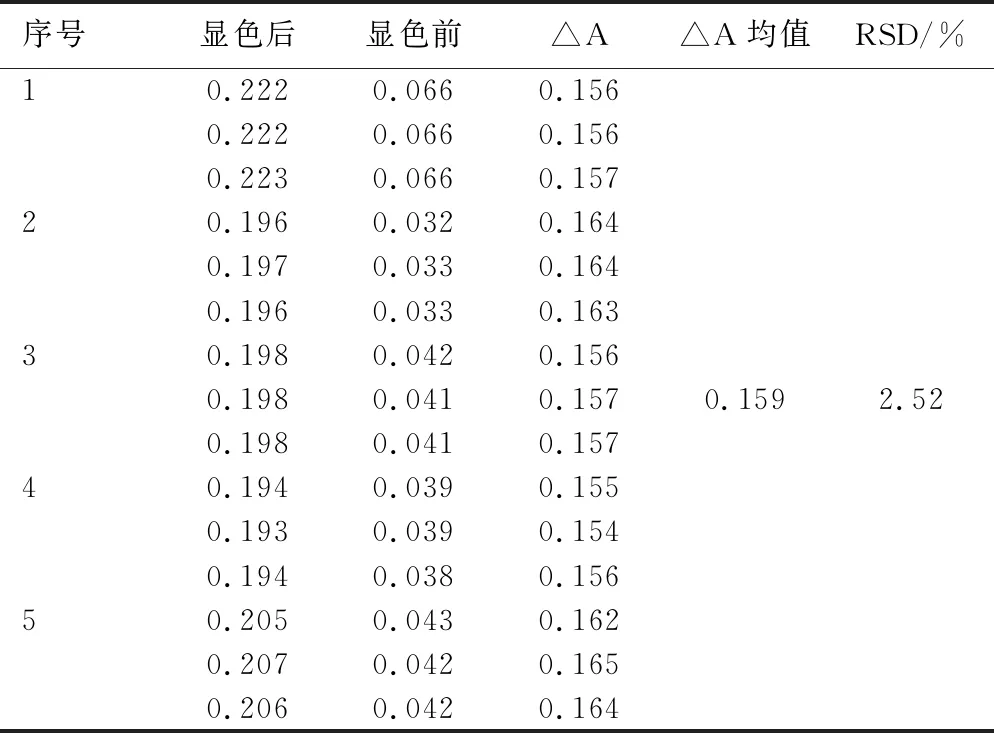
表4 重复性结果
2.1.3.4加样回收率 按照“1.3.6.4”项下的方法,得平均回收率为101.69%,回收率的RSD值为1.89%,结果表明该方法准确度良好。见表5。

表5 加样回收率结果
2.1.3.5样品含量测定 按照“1.3.6.5”项下的方法,得吸光度差△A,计算决明子中总蒽醌含量的均值为0.0550,RSD值为16.47%,结果表明不同批次决明子清炒品间总蒽醌含量有所差异。见表6。

表6 样品含量测定结果
2.2正交试验结果 按表1因素水平表的实验设计、“1.3.7”项下的炮制工艺及“1.3.8.1”项下的数据处理方法,以综合评价为数据处理指标,获得正交试验结果及方差分析结果。由表7和表8可知,各因素对综合评价的影响顺序为B>A>C,即炒制火候影响最大,其次为炒制方法,最后为炒制时间。其中,炮制方法和炮制火候对综合评价具有显著性影响。由K值可得最佳的炮制工艺为A3B3C1,即炮制方法为蛤粉炒,蛤粉炒火候为武火,炒制时间为3 min。
2.3蛤粉炒工艺的验证 按“1.3.7”项下炮制工艺和“1.3.8”项下数据处理方法,测定蛤粉炒武火3 min炮制工艺的综合评价指标的均值为0.9334,清炒武火3 min炮制工艺的综合评价指标的均值为0.7824。测定结果和独立样本t检验结果见表9。由表9可知,蛤粉炒武火3 min和清炒武火3 min两种炮制工艺的综合评价比较差异有统计学意义(P<0.05)。

表7 正交试验结果及极差分析结果
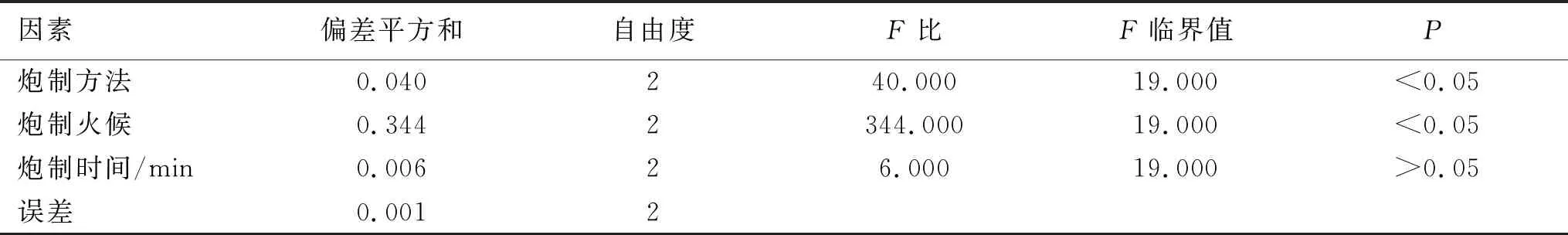
表8 正交试验方差分析结果

表9 砂炒工艺验证结果
3 讨论
康建等[10]采用直接紫外-可见分光光度法测定决明子总蒽醌的含量,该法操作相对简单,但在所选择的测定波长处未能消除其他物质的干扰,因此会影响总蒽醌的含量测定结果。鄢胜君、张小梅等[11-12]采用大孔吸附树脂、聚酰胺树脂等前处理来排除干扰物质的影响,但处理过程相对繁琐、费时。采用差示分光光度法可在所选择的测定波长处消除决明子中干扰物质的影响,准确测定决明子中总蒽醌(含游离蒽醌)的含量,操作简便,结果可靠[13-14]。研究发现,不同批次决明子清炒品间总蒽醌含量有所差异,不同产地的决明子清炒品与决明子生品间总蒽醌含量差异较为明显。本文在决明子炒制工艺考察过程中,参照有关文献[9]确定了炮制火候温度为:文火:80~120 ℃、中火:120~150 ℃、武火:150~220 ℃。待炒制温度达到规定温度范围开始炒制计时。关于红外测温枪发射频率的选择,清炒时,中药材多为植物,其发射率与木材接近为0.95[15]。砂炒时,发射频率则以砂的发射频率为准,为0.90;蛤粉炒,查阅资料可知蛤粉主要成分为碳酸钙,因此其发射频率与石灰石接近,为0.98。加辅料炒,其用量则根据《中国药典》(2020年版)确定,砂炒:以掩埋待炮制品为度;蛤粉炒:每100 kg待炮制品,用蛤粉30~50 kg[16]。
决明子炮制工艺考察结果为炒制方法为蛤粉炒,蛤粉炒工艺为武火、炒制时间为3 min。以游离蒽醌含量和总蒽醌含量的比值为评价指标,采用蛤粉炒对决明子进行炮制,与《中国药典》(2020年版)收载的决明子清炒法相比差异具有统计学意义。将三种不同炮制方法进行比较。清炒时,只有药材的一面与锅接触受
热,其受热面积很小,受热不均匀,操作不当,甚至会煳化与焦化[17];而蛤粉炒时,则以蛤粉作传热体,增大了待炮制品的受热面积而且使其受热均匀,成品性状均匀[18-19]。砂炒时,虽然能解决清炒的一些弊端,但与蛤粉炒比较,其颗粒粒径较蛤粉颗粒粒径大,减小了待炮制品的受热面积。因此蛤粉炒可更好地用于决明子的炮制。
猜你喜欢
煤炭与化工(2023年3期)2023-05-19 00:49:38
基层中医药(2021年4期)2021-07-22 07:15:44
中成药(2018年10期)2018-10-26 03:41:32
山东化工(2018年15期)2018-09-20 08:55:34
天然产物研究与开发(2018年8期)2018-09-10 05:48:22
中成药(2018年4期)2018-04-26 07:12:43
中成药(2018年4期)2018-04-26 07:12:34
中成药(2017年4期)2017-05-17 06:09:36
首都食品与医药(2015年18期)2015-11-03 05:59:08
中国当代医药(2015年23期)2015-03-01 02:05:37
