
UDG在两种水生动物病毒核酸片段检测中的抗污染效果研究
2022-03-28 12:43丁能水李奕雅汪小东许弟群
闽南师范大学学报(自然科学版) 2022年1期
陈 凡,丁能水,李奕雅,汪小东,许弟群
(1.闽南师范大学生物科学与技术学院,福建 漳州 363000;2.福建傲农生物科技集团股份有限公司,福建 漳州 363000;3.闽南师范大学体育学院,福建 漳州 363000)
聚合酶链式反应(polymerase cain reaction,PCR)是一种指数级扩增微量基因片段的核酸检测技术,拥有极高的灵敏度,被广泛应用于医学检测、环境监测、转基因食品、水产养殖等多个领域.以常规PCR技术为基础,衍生出诸如多重PCR(Multiplex PCR)、实时荧光定量PCR(qPCR)、巢式PCR(Nested PCR)等技术,又进一步扩大了PCR 技术的应用范围[1].自2020年初新冠疫情爆发以来,基于PCR 技术的“核酸检测”,更是作为确诊COVID-19 的“金标准”,为人们所熟知[2].实际应用中,由于PCR 及其衍生技术具有极高的灵敏度,故其反应过程特别是试剂混合与加样步骤特别容易受到污染物(如阳性DNA 气溶胶等)的干扰,得出假阳性结果.且假阳性的产生原因极难排查,更难以彻底解决,长期以来不断困扰着广大检验人员和科技工作者[3-4].因此,实验室环境中的污染物控制一直是相关检测稳定性的重要影响因素.
一般情况下,若PCR 反应体系中存在dUTP,则dUTP 能与dTTP 产生竞争,掺入扩增产物中,形成带有尿嘧啶(U)的DNA 分子.而尿嘧啶-DNA 糖基化酶(Uracil-DNA Glycosylase,UDG)能特异性地切断DNA 分子中尿嘧啶碱基与脱氧核糖间的N-糖苷键,移去尿嘧啶,生成无碱基位点,在DNA 片段上挖开缺口[5].带有缺口的DNA在随后的PCR循环中,将无法作为完整的模板被扩增.基于这一特性,UDG被用于PCR反应中的污染控制,且效果得到了肯定[6-10].
近年来,PCR 预混液试剂盒得到普遍应用,使用时仅需加入适量引物、模板和水即可进行PCR 扩增,简化了操作,也减少了操作过程中可能导致的污染.因此,在常规和定量PCR 实验中,预混试剂盒已经占据了主导地位.然而,此类试剂盒中的酶和dNTP 以预混方式存在,浓度不可自由调节,若要向其中添加dUTP,实现抗污染效果,则与传统试剂盒相比,其反应条件的优化更加困难.实际使用中,若体系中dUTP含量过高,则DNA 聚合酶活性将受到抑制,导致扩增效率(检测灵敏度)降低甚至完全无法扩增模板;若dUTP 含量过低,则不能保证PCR 产物中掺入足量的尿嘧啶,对UDG 活性的发挥不利[10].因此,本研究以PCR产物中A-T占比存在较大差异的鲤鱼浮肿病病毒(CEV)和罗非鱼湖病病毒(TiLV)核酸检测为例,对使用PCR 预混液时,dUTP 的添加量及对应条件下UDG 的去污染效果进行了评价研究,以期为该条件下UDG抗污染手段的应用和优化提供实验设计和使用效果的量化参考.
1 材料与方法
1.1 感受态细胞、工具酶与试剂
化学感受态细胞DH5α(E.coliDH5α,转化效率>5×108cfu/μg)购于上海唯地生物技术有限公司.San-Prep柱式质粒DNA小量抽提试剂盒,4S Red Plus核酸染色剂,DNA分子量标准Marker(100~5 000 bp),琼脂糖,SanTaq Plus PCR 扩增试剂盒(下文简称“SanTaq”),SGExcel UltraSYBR Master 预混液,SGExcel GoldStar TaqMan Master 预混液等,购于生工生物工程(上海)股份有限公司.2×Rapid Taq Master Mix(下文简称“RapidTaq”),E.coliUDG(5 U/μL),dUTP(100 mM)等,购于南京诺唯赞生物科技股份有限公司.2×HLingene PCR Master Mix(下文简称“HLTaq”)购于上海惠凌生物技术有限公司.引物合成与质粒测序委托苏州金唯智生物科技有限公司进行.若无特殊说明,各试剂使用时均严格按照对应的说明书要求进行操作或设置反应条件.
1.2 阳性质粒与引物
阳性质粒pUC19-CEV和pUC19-TiLV均由本课题组构建并保存.CEV阳性质粒包含鲤鱼浮肿病病毒的P4a 蛋白基因保守序列以及5′UTR 序列[11],TiLV 阳性质粒包含罗非鱼湖病病毒基因的保守序列[12].质粒结构及检测中使用的引物结合位点与扩增片段长度如图1所示,对应的引物序列如表1所示.质粒提取过程严格按照抽提试剂盒说明书进行.以TE缓冲液溶解纯化后的质粒分子,并采用NanoDrop 2000c超微量分光光度计(thermo fisher)测定其浓度.

表1 本研究使用的引物Tab.1 Primers used in this study

图1 本研究使用的阳性质粒结构及对应的引物结合位点示意图Fig.1 Schematic representation of the positive plasmid structures and corresponding primer binding sites used in this study
1.3 PCR条件与步骤
本研究使用的PCR 反应条件为,(1)常规PCR(SanTaq、HLTaq、RapidTaq):95 ℃(120 s)→95 ℃(15 s)→55 ℃(15 s)→72 ℃(SanTaq 和HLTaq 为1 000 bp/min,RapidTaq 固定为5 s,35 循环)→72 ℃(120 s)→10 ℃(Hold);(2)染料法qPCR(SGExcel UltraSYBR):95 ℃(180 s)→95 ℃(15 s)→55 ℃(20 s)→72 ℃(30 s,40循环);(3)探针法qPCR(SGExcel GoldStar TaqMan):95 ℃(180 s)→95 ℃(20 s)→60 ℃(30 s,40循环).其中常规PCR 使用GE9612T-S 基因扩增仪(杭州柏恒科技有限公司)完成,荧光定量PCR 使用QuantStudio™6实时荧光定量PCR系统(Applied Biosystems)完成.
巢式PCR步骤,(1)CEV:使用“BF+BR”引物组扩增模板中的528 bp片段,经琼脂糖凝胶电泳确认能够扩增出特异性DNA 条带的样品,以TE 缓冲液将第一轮扩增产物稀释1 000 倍,作为第二轮反应的模板.再使用“IF+IR”扩增第二轮模板中的478 bp 片段,并通过琼脂糖凝胶电泳(1.5%,下同)检测特异性条带.若第一轮扩增后无法检测到特异性条带,则直接使用该产物(不稀释)作为模板进行第二轮扩增并检测.(2)TiLV:除引物外,检测与模板准备步骤与CEV一致,第一轮使用引物组“Nested ext-1+ME1”,扩增产物共415 bp;第二轮使用引物组“ME1+ME2”,扩增产物共250 bp.
荧光定量PCR:取待测DNA 作为模板,直接通过SYBR Green 染料法或TaqMan 探针法进行定量检测,每个样品使用3个复孔.此外,为便于数据比较并提高实验的稳定性,在各标准曲线的建立过程中,均以全长质粒作为扩增模板.
1.4 Taq酶对dUTP的耐受性检验
向PCR反应体系中添加不同浓度的dUTP溶液,形成特定的dUTP浓度梯度,再利用各PCR预混液对pUC19-CEV 和pUC19-TiLV 质粒的巢式第一轮阳性片段进行扩增,并利用琼脂糖凝胶电泳检测扩增结果,以确认dUTP浓度对PCR反应的影响.
1.5 UDG反应条件
按照特定的实验设计,将同一扩增条件下得到的PCR 产物分装若干份,并向每份PCR 产物中添加一定量的UDG,对反应体系进行漩涡混匀,再将混合液以2 000 g离心15 s,随后转移到水浴锅或PCR 仪内,37 ℃孵育10 min,使UDG 充分发挥作用,降解底物.最后将孵育温度提升至95 ℃,维持2 min,对UDG 进行灭活处理.经UDG 处理后的PCR 产物,可通过琼脂糖凝胶电泳进行半定量评价,或以TE 缓冲液稀释1 000倍后,以qPCR法对降解效果进行定量评价.
1.6 UDG对模板DNA降解效果的评价
电泳法:使用PCR预混液以及不同的巢式引物对CEV和TiLV阳性片段进行扩增.随后参照方法1.5,使用不同的UDG浓度对扩增产物进行处理.再对处理后的PCR产物进行琼脂糖凝胶电泳检测.
染料法qPCR:使用特定PCR 预混液和引物组扩增pUC19-CEV(扩增用引物组为“BF+BR”)或pUC19-TiLV(扩增用引物组为“Nested-ext1+ME1”)质粒中的阳性DNA 片段,再采用不同浓度的UDG 对PCR产物进行降解,最后将降解产物以TE缓冲液稀释1 000倍,作为qPCR模板,配合巢式引物上机检测.检测时使用基于SYBR Green的荧光染料和ROX参比作为荧光定量依据.
探针法qPCR:使用特定的PCR 预混液和引物组扩增pUC19-CEV(扩增用引物组为“BF+BR”)或pUC19-TiLV(扩增用引物组为“SF+SR”)中的阳性DNA 片段,并采用不同浓度的UDG 对PCR 产物进行降解,再将降解产物以TE缓冲液稀释1 000倍后,作为qPCR模板,配合探针法引物上机检测.检测时使用基于FAM-BHQ1的探针和ROX参比作为荧光定量依据.
1.7 统计学处理
所有数据使用Minitab v17.0 软件进行数值统计.荧光定量PCR 复孔检测结果的Ct 值以“平均值±标准差”表示(由于qPCR 数据的平均值远大于标准差,使各检测结果的CV 值均小于1%,故图5-6 不对标准差进行绘制),利用线性回归得到不同模板添加量对应的回归方程与决定系数后,根据回归方程反推待测样品中的模板含量并进行相互比较与计算.
2 结果与讨论
2.1 Taq酶对dUTP的耐受性分析
不同Taq酶对dUTP作为反应底物的耐受性实验结果如图2所示.由图中的电泳条带可见:SanTaq和HLTaq的扩增产量均随着反应体系中dUTP浓度的增加而呈现降低趋势.更为明显的是,RapidTaq对体系中dUTP 的存在完全不耐受,仅向反应体系中添加100 μmol/L 的dUTP 即可完全抑制其活性.因此,后续实验不再以RapidTaq为实验材料展开进一步探讨.反观SanTaq和HLTaq对应的实验结果,可以发现:在检测CEV基因时,若dUTP浓度达到400 μmol/L,则SanTaq的扩增效率显著降低;若dUTP浓度达到800 μmol/L,则HLTaq 的扩增性能也有明显下降.而检测TiLV 基因时,dUTP 对两种酶特别是HLTaq 扩增性能的影响则相对较小.这一现象产生的原因可能在于:CEV模板中A-T碱基的占比为68.4%,远高于TiLV的50.6%(图1),导致CEV模板扩增过程中Taq酶与dUTP的相互作用更为频繁.
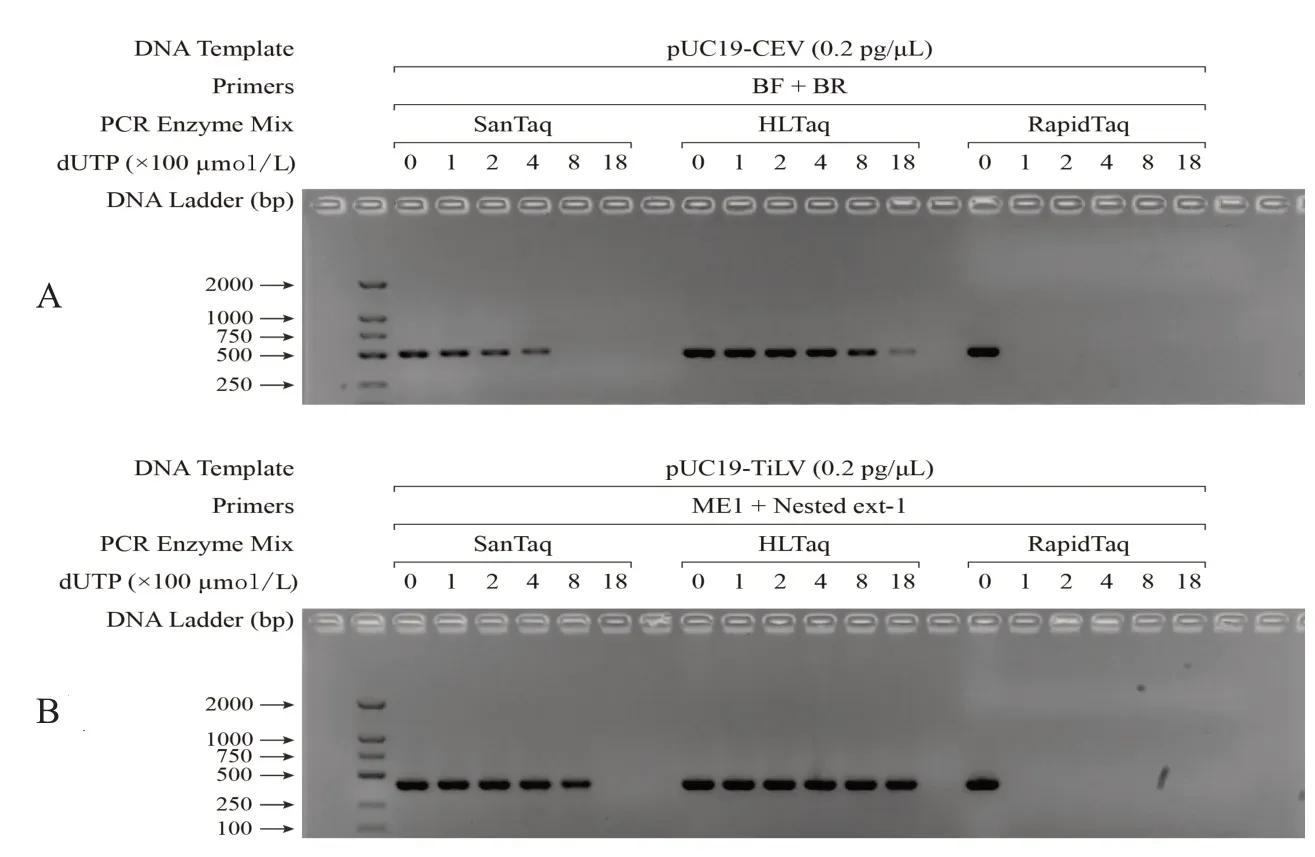
图2 三种Taq酶对dUTP的耐受性实验结果Fig.2 Experiment results of the dUTP tolerance with three Taq DNA polymerase
为确保PCR 体系中加入的dUTP 不对扩增效率产生较大干扰,本研究以近年来参与全国水生动物防疫系统实验室检测能力验证的检验数据为基础,计算得到:标准核酸抽提手段下,病鱼样本中能够提取到的CEV阳性DNA模板浓度最小值约等价于pUC19-CEV质粒0.022 pg/μL;病鱼中的TiLV阳性RNA经逆转录后,对应的DNA模板浓度最小值约等价于pUC19-TiLV质粒0.007 pg/μL.因此,本研究考察了阳性质粒用量0.2 pg/μL 以及0.002 pg/μL 情况下,dUTP 浓度梯度对两种Taq 酶扩增效果的影响,结果(图3)表明:(1)dUTP用量不超过200 μmol/L时,两种酶在高低两种模板浓度下的扩增效果受dUTP影响较小;(2)dUTP用量不超过800 μmol/L时,即使在低模板浓度下,两种酶也能通过PCR扩增准确判断阳性模板的存在.其中,HLTaq 在dUTP 浓度达到1 800 μmol/L的情况下,对低浓度模板依然有效.为便于横向比较,后续统一以200 μmol/L的dUTP浓度为基本反应条件,展开进一步研究.
2.2 UDG用量对模板降解效果的影响(电泳法)
UDG 用量对模板降解效果的影响如图4所示.通过对图4A-D 组结果的分析,可知:(1)UDG 对不含U 的底物不产生降解,可认为UDG 在本研究使用的反应条件下无非特异活性;(2)不同UDG 浓度对含U的底物均产生了极其显著的降解效果,且可以认为:随着UDG 浓度的升高,降解效果愈发理想(降解产物的分子量均一性逐渐提高).但对比图中展示的各电泳结果,也能看出:UDG 用量超过0.2 U/10 μL 时(即试剂说明书推荐的最低用量),降解效果未随用量的提高而发生显著变化.

图4 不同UDG浓度对各PCR产物的降解效果Fig.4 Amplicon degradation by UDG concentration gradient
2.3 UDG用量对模板降解效果的影响(染料法qPCR)
以qPCR(染料法)对UDG的降解效果进行定量评价,结果如图5所示(因同组实验中任意两个数据均存在显著差异,故图中不再额外标记).当使用0.05、0.2、1 U/10 μL 三种浓度的UDG 处理同一PCR 产物时,不论扩增模板是CEV 还是TiLV 阳性片段,也不论使用了何种预混液,UDG 均表现出良好的降解效果.仅在0.05 U/10 μL的用量下,其模板的平均降解水平就可达99.9%左右;若提高UDG用量至1 U/10 μL,则平均降解水平可上升至99.98%左右.
从本组结果可推导如下结论:(1)UDG 对几种含U 模板均有显著降解效果,若使用0.2 U/10 μL 的UDG,则降解效率通常在99.95%以上.(2)从降解效率分析,与0.2 U/10 μL 的UDG 浓度相比,若仅使用0.05 U/10 μL,其作用效果明显下降;若使用1 U/10 μL 的UDG 浓度,则效率提高有限或不提高.(3)使用SanTaq 扩增的产物,其降解效率普遍高于HLTaq,其原因可能在于:1)同条件下,SanTaq 可能更容易将dUTP 掺入终产物,有利于UDG 发挥作用;2)SanTaq反应体系的配方(如缓冲液浓度、pH 值等)对UDG 的活性更为友好;3)SanTaq扩增效率较HLTaq更低,在酶活相同的情况下,更低的底物浓度对反应完全更为有利.但具体由哪个因素主导,仍有待进一步研究.
2.4 UDG用量对模板降解效果的影响(探针法qPCR)
以qPCR(探针法)对UDG的降解效果进行定量评价,其实验结果如图6所示(因同组实验中任意两个数据均存在显著差异,故图中不再额外标记),当使用由低到高三种UDG浓度处理同一PCR产物时,其结果与图5相似——不同的UDG浓度均能在本组实验中展现出对模板的降解效果.
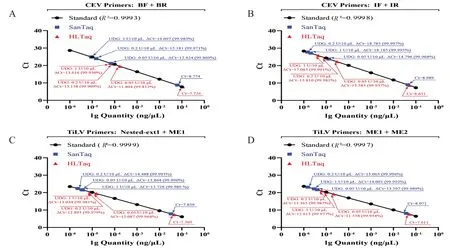
图5 不同UDG浓度对不同PCR产物降解效果的定量分析(qPCR染料法)Fig.5 SYBR Green based quantitative evaluation of amplicon degradation by UDG concentration gradient

图6 不同UDG浓度对不同PCR产物降解效果的定量分析(qPCR探针法)Fig.6 TaqMan based quantitative evaluation of amplicon degradation by UDG concentration gradient
与染料法qPCR检测不同的是:利用qPCR(探针法)检测特定目标时,为保证扩增效率,通常要求扩增的片段小于200 bp[16].但更短的片段意味着更低的尿嘧啶掺入概率,势必对UDG的作用效果产生影响.因此,本组实验的结果明显可见:(1)与图5所示数据相比,当扩增产物较短时(探针法CEV 产物为76 bp,TiLV产物为67 bp),UDG对潜在污染物的降解效率有明显下降.即便使用较高的UDG浓度,其CEV模板降解效率也只能达到99.9%左右,相比染料法的结果降低了约10 倍,且这一现象在TiLV 模板的降解中(最高降解率仅98.438%)更为明显.因此,可认为过短的产物对UDG的降解作用不利.(2)使用探针法检测时,CEV模板(A-T:65.8%)降解效率明显高于TiLV模板(A-T:50.7%),可见底物的A-T含量对UDG活性的影响很大,含量越高则UDG 作用效果越好.(3)本组实验数据显示HLTaq 扩增产物的降解效率高于SanTaq,与图5所示结论相反.推测其原因可能在于:探针法检测过程中,荧光信号的强弱不单取决于扩增反应本身,探针分子与模板的结合能力也至关重要.且探针分子只能选择性地结合DNA双链中的某一链,故探针分子中腺嘌呤(A,能够与模板中的T或U配对)的数量与分布就可能对反应过程中的荧光信号产生较大影响.而通过分析CEV 探针(qProbe)和TiLV 探针(TiLV-P1)的序列,可以发现其中A 的占比均小于整个阳性模板的均值.特别是TiLV-P1 中,A 占比仅5.6%,远低于探针法扩增区域中的20.9%,故检测时可能呈现出与染料法不一致的结果.因此,在UDG 对底物的降解效率评价中,染料法的实验结果可能更具有参考价值.(4)HLTaq扩增产物,在UDG用量为0.05 U/10 μL时,降解效率远不及0.2 U/10 μL以上的条件,也从另一方面佐证了SanTaq的反应体系配方对UDG活性的表达更为有利,即使用HLTaq扩增检测模板时,需使用较高(过量)的UDG浓度,才能达到理想的效果.
3 结语
UDG 属于生物进化过程中较为保守的DNA 修复酶系,是由六个亚家族组成的蛋白质超家族.除了其自身独特的生物学功能外,在PCR反应中,UDG通常被用于DNA残留污染的清除.
(1)UDG 的实际作用效果受到扩增产物大小、产物中A-T 碱基比例,以及Taq 酶自身对反应体系中dUTP浓度耐受的影响.总体而言,扩增产物越长,产物中A-T碱基对越多,Taq对dUTP耐受性越好的情况下,越有利于UDG 的抗污染作用.故使用UDG 作为抗污染试剂前,建议根据具体实验材料,首先进行预实验,摸索得到较好的反应条件后,再铺开后续的研究或检测工作.
(2)使用SanTaq 和HLTaq 两种PCR 预混液试剂盒作为检测工具时,向反应体系中添加200 μmol/L 的dUTP,并使用0.2 U/10 μL 的UDG 浓度清除潜在阳性污染物,是一种比较适宜的条件(仅使用HLTaq,则dUTP浓度可进一步提高).在这一条件下,若进行常规PCR,则UDG对污染物的清除效率在99.9%以上,且多数情况高于99.95%.若实验环境中仅存在轻度污染,UDG 能够较好地发挥作用.若使用qPCR(探针法)作为检测手段,则同样条件下污染物的清除率只能达到96.5%以上,对于高灵敏度的qPCR体系,这一清除率则略显不足.但值得注意的是:研究中使用了阳性片段的降解产物作为模板进行定量评价.事实上,此类降解产物中仍不乏能够正常结合引物或探针的片段,只是模板断裂后扩增效率大幅下降而已,但其仍具备诱导产生荧光信号的能力.故可认为:本研究中UDG 的降解效率实际上优于qPCR 法的定量检测结果.若需要更为准确地评价UDG的作用效果,则必须借助数字PCR等更为先进的检测工具.
(3)鉴于UDG 的作用特点,若污染物不含或少含尿嘧啶碱基,则其难以发挥作用.因此,在实际操作中,其抗污染能力是有一定限度的,操作人员除了添加dUTP和UDG作为抗污染手段外,更应该注意养成良好的操作习惯,并注意加强实验室标准化管理,才能确保检测的准确度.
本研究系统地分析了以PCR预混液试剂盒为检测工具时,UDG 酶在CEV 和TiLV 检测中的抗污染作用,一方面总结得出了检测时较为适宜的dUTP和UDG 用量,另一方面也为特定条件下UDG 对污染性模板的清除率做了定量统计,能为该技术的具体应用与进一步优化提供实验依据.
猜你喜欢
中学生物学(2022年8期)2022-10-13
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
三农资讯半月报(2020年11期)2020-06-21
当代化工(2020年2期)2020-03-18
江苏农业学报(2019年1期)2019-09-10
小雪花·初中高分作文(2017年10期)2018-05-15
今日财富(2017年32期)2017-10-19
卷宗(2014年7期)2014-08-27
卷宗(2014年1期)2014-03-20
中学生物学(2008年12期)2008-12-27
