
在线净化/液相色谱-四极杆/静电场轨道阱高分辨质谱法快速筛查水产品中123种兽药残留
2022-03-22 06:40欧阳少伦陈文锐安文佳庄燕君
分析测试学报 2022年3期
欧阳少伦,蓝 草,陈文锐,安文佳,林 峰,庄燕君
(广州海关技术中心,广东 广州 510623)
随着工业化的迅速发展,工业污水和环境污染日益严重,加之一些地区养殖密度过高,饲料质量差,致使水生生物抵抗力低、发病率增加。为了治疗和预防各种病害、提高产量效益,水产品的养殖过程中存在误用、滥用各种药物的情况,主要包括三苯甲烷类染料、四环素类、氯霉素类、磺胺类、喹诺酮类等。人们长期食用含渔药残留的水产品,药物易在人体内蓄积,可能会产生致畸、致癌的严重危害[1],有些渔药残留会间接增强人体病原菌的耐药性,增加治疗难度。因此,世界各国对水产品中的兽药残留限量做了严格规定[2]。
目前对水产品中兽药残留的检测方法主要以气相色谱法(GC)[3-4]、液相色谱法(HPLC)[5-6]、气相色谱-质谱法(GC-MS)[7-8]、液相色谱-三重四极杆质谱联用法(LC-MS/MS)[9-12]、液相色谱-飞行时间质谱法(LC-TOF MS)[13-14]和液相色谱-四极杆/静电场轨道阱高分辨质谱法(LC-HRMS)[15-16]为主。但现有研究检测的药物种类单一,前处理净化步骤较多,高通量检测仍具有一定的局限性。水产品中兽药残留检测已趋向多种类、高通量发展[17],目前已有文献报道了牛奶[18-19]、饲料[20]、猪肉[21]等基质中兽药残留的测定方法,但在水产品中兽药多残留高通量快速筛查方面的研究鲜有报道。
在线净化技术TurboFlow主要通过扩散溶解、体积排阻等手段达到净化目的,比传统的固相萃取和基质分散萃取更具优势,已在食品领域得到了较好的应用[22-23]。本文建立了在线净化/液相色谱-四极杆/静电场轨道阱高分辨质谱高通量筛查水产品中123种兽药残留的方法,包括20种磺胺类、6种大环内酯类、16种喹诺酮类、12种β-激动剂、17种苯并咪唑类、4种四环素类、2种镇静剂、6种激素、4种β-内酰胺类、1种氨苯砜、2种抗球虫药、2种染料、24种糖皮质激素、1种氯霉素、6种雷索酸内酯类。该方法前处理简单,结果准确,适用于大批量水产品中多种兽药残留的快速筛查。
1 实验部分
1.1 试剂与仪器
乙腈、甲醇、丙酮、异丙醇(HPLC级,Fisher Scientific公司);甲酸(分析纯,Merck公司);所用试剂除注明外均为分析纯,实验用水为超纯水。123种化合物的标准品购自德国Dr.Ehrenstorfer公司、德国Witega公司、美国Sigma公司、北京振翔公司、中国食品药品检定研究院、上海安谱公司,纯度为91.2%~99.9%。
Transcend TLX在线净化液相色谱Q-Exactive静电场轨道阱质谱分析系统(由双流路大体积大容量自动进样器、1个低压四元泵、1个高压二元泵和Q-Exactive-Orbitrap质谱仪组成,美国Thermo Fisher公司)。Milli-Q超纯水器(美国Millipore公司);CPA22SD分析天平、BT 124S分析天平(Sartorius公司);SiGMA 1-14高速离心机(德国Sigma公司);MS3 basic旋涡振荡器(IKA公司);Promax 2020水平振荡器(德国Heidolph公司);S300H超声波水浴(德国Elma公司)。
数据处理软件:TraceFinder EFS、Xcalibur、Aria均为美国Thermo Fisher Scientific公司产品。
1.2 标准溶液配制
称取10 mg标准品,分别用甲醇、乙腈或水等溶剂溶解并定容至10 mL,配制为1 mg/mL的单标储备液,置于-18℃保存。使用时根据需要将化合物分组用甲醇进行混合或稀释,所有稀释液均置于4℃下避光保存。
1.3 仪器条件
1.3.1色谱条件在线净化程序主要为净化和分离过程,净化过程(TFC)由上样泵驱动净化流动相在TurboFlow柱上完成,分离过程(LC)由分离泵驱动分析流动相在分析柱上完成。整个程序通过切换2个六通阀使流路改变,实现在线净化富集。
TurboFlow净化柱:CycloneTMP(0.5 mm×50 mm,Thermo Fisher Scientific公司),净化泵流动相:A为0.1%甲酸水溶液,B为甲醇,C为乙腈,D为丙酮∶异丙醇∶乙腈(20∶40∶40,体积比)。柱温:室温,进样量:50μL。
色谱柱:Atlantis T3(100 mm×4.6 mm,3μm,Waters公司);柱温:35℃。流动相:正离子检测和负离子检测(糖皮质激素)时,A为0.1%甲酸水溶液,B为乙腈;负离子检测(除糖皮质激素外)时,A为水,B为乙腈。梯度洗脱程序见表1。

表1 在线净化梯度洗脱程序Table 1 Gradient elution procedure of TurboFlow on-line cleanup
1.3.2质谱条件Q-Exactive-Orbitrap质谱仪(美国Thermo Fisher公司),配有电喷雾离子源(ESI)。正或负离子化模式;扫描范围:m/z100~1 000;一级全扫描分辨率:R=70 000;喷雾电压:3 500 V;鞘气流速:40 arb;辅助气流速:7 arb;毛细管温度:320℃;包含列表:on;离子最大容纳数量:1e6;离子最大注入时间:100 ms;二级离子全扫描分辨率:17 500;归一化碰撞能量:20%、40%、60%。其他质谱信息见表2。
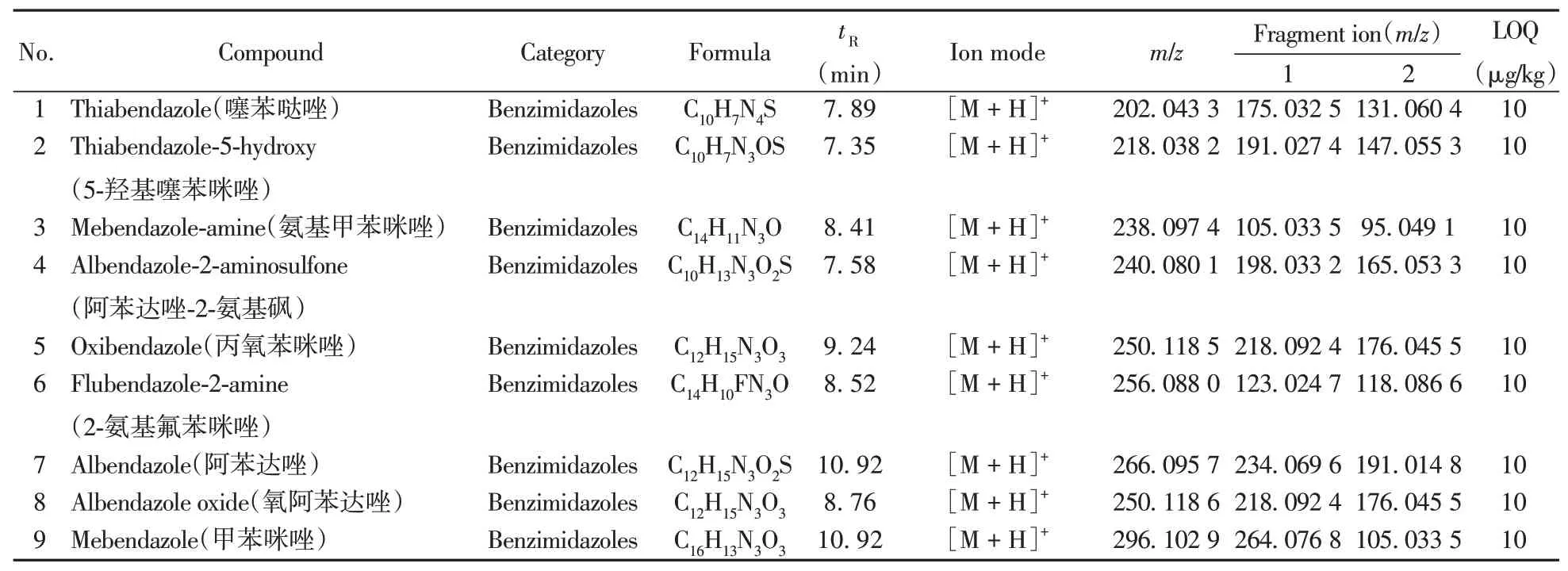
表2 123种兽药的名称、分子式、部分质谱参数和定量下限Table 2 Names,formulas,some MS parameters and LOQs of 123 veterinary drugs
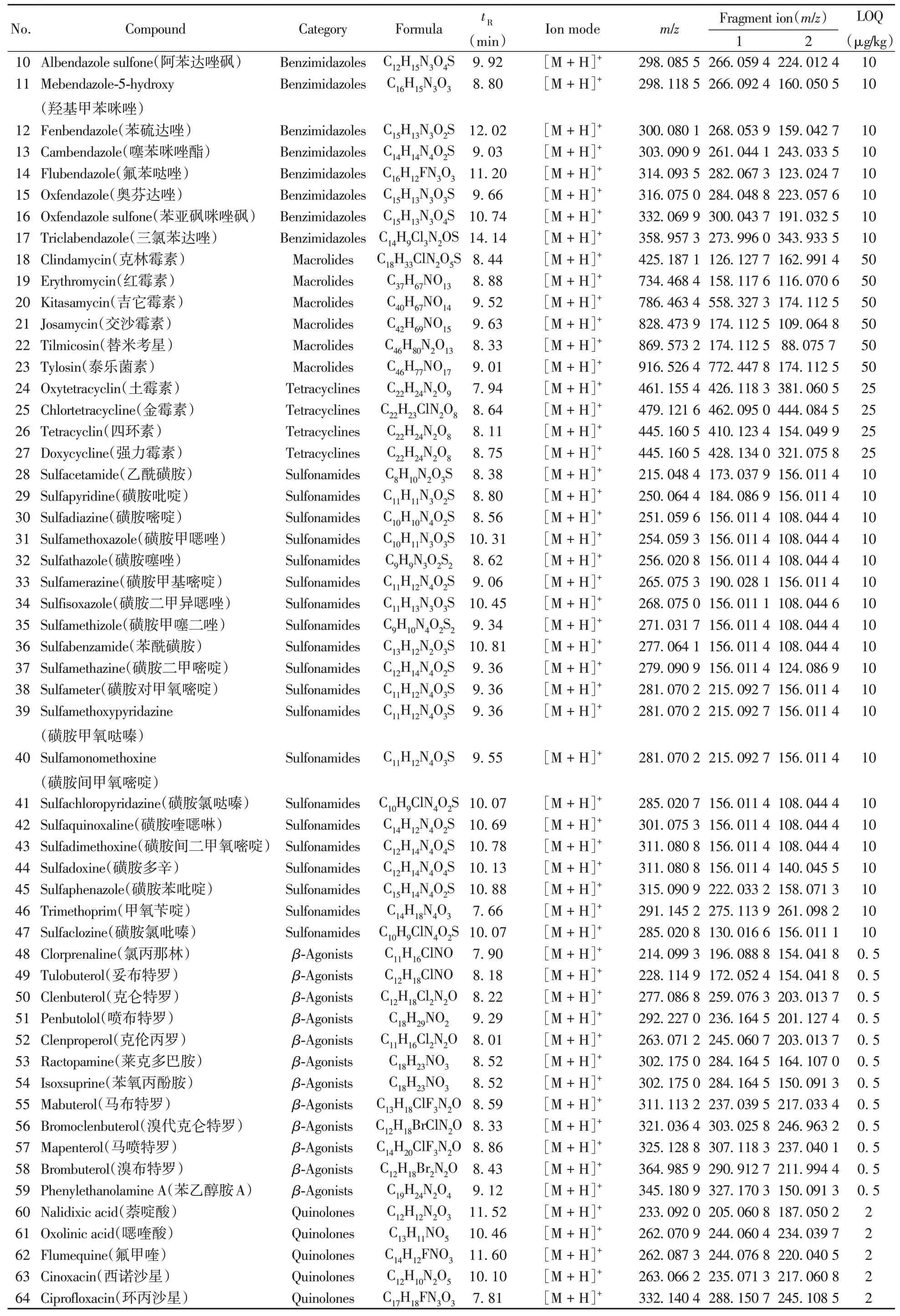
(续表2)

(续表2)

(续表2)
1.4 样品前处理
称取5 g均质样品(精确至0.01 g)于50 mL离心管中,加入7.5 mL乙腈-水(8∶2,体积比,下同)溶液,涡旋1 min,超声15 min,水平振荡15 min,以4 500 r/min离心5 min,上清液转移至50 mL离心管中;残渣再加入7.5 mL乙腈-水(8∶2)溶液,用玻棒捣碎下层残渣,水平振荡15 min,以4 500 r/min离心5 min,上清液合并于50 mL离心管中混匀,取适量样液经0.2μm滤膜过滤后上机测定。
2 结果与讨论
2.1 提取条件的优化
实验对比了乙腈-水(8∶2)溶液、乙腈和1%乙酸乙腈3种提取溶剂对目标化合物的提取效率。结果表明,乙腈对大部分目标分析物的提取效果好,且能有效地沉淀样品中的蛋白质,但部分目标分析物(如β-内酰胺类和四环素类药物)的极性较大,提取剂中需加入水才能得到较好的提取效率;而喹诺酮类药物在弱酸性条件下的提取效率更高。对于苯并咪唑类、磺胺类、大环内酯类等兽药,上述3种提取溶剂均有很好的提取效率,其中乙腈-水(8∶2)溶液的提取效率略高;倍他米松、地塞米松、可的松醋酸醋和泼尼松龙醋酸酯在乙腈中的提取效率很低,可的松醋酸酯和泼尼松龙醋酸酯采用1%乙酸乙腈时的提取效率极低;对于喷布特罗、莱克多巴胺、苯氧丙酚胺、马布特罗、溴代克仑特罗、马喷特罗、溴布特罗、四环素类,以乙腈为提取溶剂时的提取效率很低,乙腈-水(8∶2)溶液的提取效率略高于1%乙酸乙腈。综合考虑目标分析物的检测浓度水平和提取效率,本实验采用乙腈-水(8∶2)溶液作为提取溶剂。
2.2 净化条件的优化
水产品本底复杂,会带入脂肪、类脂及其它干扰组分。目前的在线净化技术主要分为两类:一类是简单的SPE柱,可去除部分样品杂质,但不能有效去除脂类物质,需要额外的离线去除脂肪步骤;另一类是在线净化,净化柱可同时去除脂肪等类脂类物质,样品无需额外的离线去除脂肪步骤。本研究采用的TurboFlow技术属于第二类在线净化技术,流路示意图见图1。
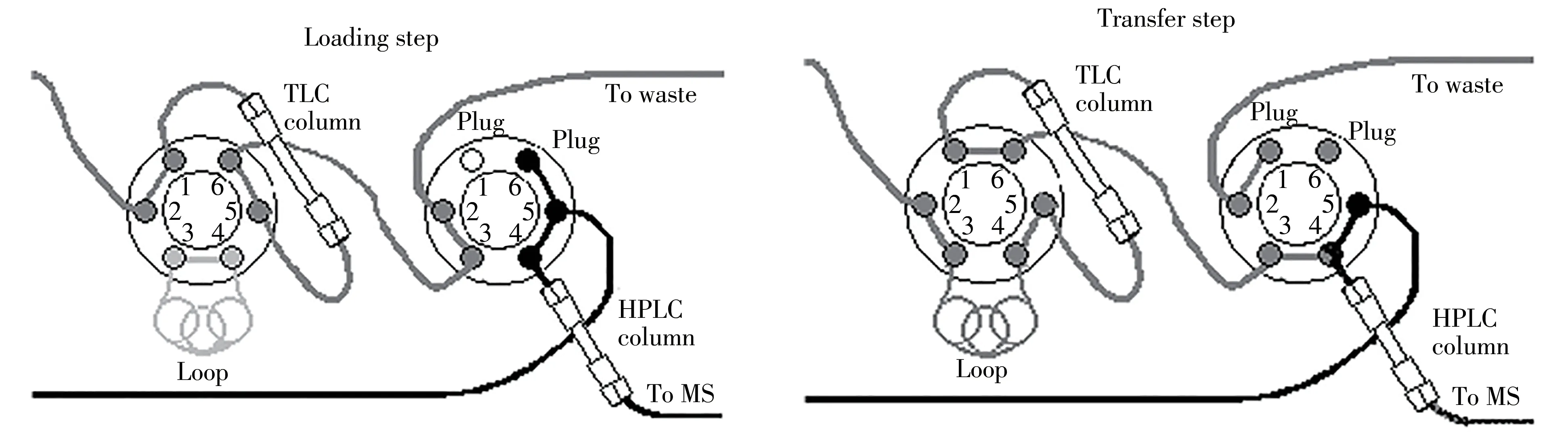
图1 TurboFlow整体流路示意图Fig.1 Schematic diagram of overall velocity path of TurboFlow
TurboFlow色谱柱由50~100μm大粒径填料填充而成,具有体积排阻作用和化学作用力。当使用大流速时(如2 mL/min),流动相在色谱柱内形成湍流,在高流速下小分子化合物有足够的时间快速扩散进入柱填料颗粒表面的小孔中,通过与填料表面的键合相相互作用保留在色谱柱上;保留较弱的小分子干扰物排出色谱系统(例如磷脂、盐类、糖类),而大分子化合物(主要为蛋白)由于扩散速度慢而没有足够的时间扩散到填料表面或内部,故不能保留在色谱柱上而被直接冲至废液中。TurboFlow技术可实现一次性去除样品中的磷脂、盐类、蛋白等干扰物,实现快速高效的分离净化。其在线净化过程主要包括上样、洗脱转移和条件化3个步骤。本研究分别对TurboFlow的上柱条件、洗脱条件和净化柱类型进行了优化。
2.2.1上柱条件的优化采用不接分析柱,分流进质谱方式优化上柱条件。分别考察了0.1%甲酸水溶液、0.1%甲酸水溶液-乙腈(98∶2)、0.1%甲酸水溶液-乙腈(95∶5)、0.1%甲酸水溶液-乙腈(90∶10)、0.1%甲酸水溶液-乙腈(75∶25)5种上柱溶剂对分析物保留的影响。结果表明:当有机相比例增至2%时,分析物在上柱步骤有不同程度的损失,随着有机相比例的增加,分析物的损失不断增大,在净化柱的保留程度越低。因此,实验采用0.1%甲酸水溶液作为上柱溶剂。
2.2.2洗脱条件的优化采用0.1%甲酸水溶液分别配制25%乙腈、50%乙腈、75%乙腈、100%乙腈、25%甲醇、50%甲醇、75%甲醇和100%甲醇作为洗脱溶剂,考察了各类代表性药物在不同洗脱溶剂下的回收率。结果表明,当洗脱溶剂的有机相比例高于水相时,由于有机溶剂比例过高导致溶剂不匹配,大部分物质的色谱峰前延严重或开叉。比较有机溶剂比例较低的4个洗脱条件发现,除了噻苯哒唑、克林霉素、乙酰磺胺、磺胺间甲氧嘧啶和氯羟吡啶等兽药,乙腈对其他大部分目标分析物的洗脱能力优于甲醇。除了糖皮质激素类,25%乙腈对大部分目标分析物的洗脱效果优于50%乙腈。综合考虑,选择25%乙腈作为洗脱溶剂。
2.2.3在线净化柱的选择由于目标分析物大部分为极性或中等极性,实验对比了反相苯基键合硅胶CycloneTMP柱(0.5 mm×50 mm)和键合硅胶C18净化柱(0.5 mm×50 mm)对目标分析物的回收率。采用样品空白基质配制质量浓度为10μg/L的混合标准溶液上样,基质匹配标准曲线进行定量,部分代表性化合物的回收率见图2。结果表明,多数目标分析物在CycloneTMP柱上的保留更好。综合考虑,选择CycloneTMP作为净化柱。
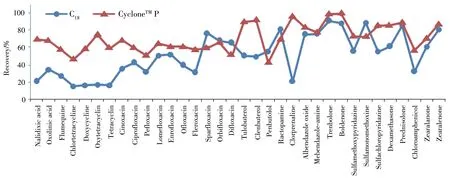
图2 代表性药物在CycloneTM P和C18柱上的回收率Fig.2 Recoveries of representative drugs on Cyclone TM P and C18 columns
2.3 药物的定性分析
采用Full Scan/ddMS2模式对123种化合物进行扫描,获得目标物的保留时间、母离子及二级质谱图等信息。采用mzVault软件建立标准二级谱库,每种目标物包含多个特征离子,结合化合物的结构、裂解原理选择相对丰度较高的2个离子作为特征碎片离子,见表2。通过Exactfinder软件导入包含化合物名称、分子式、精确质量数、保留时间和特征碎片的表格,并通过Librarymanager软件导入二级质谱图,建立了定性筛查数据库。设置筛查参数要求提取母离子精确质量数与理论质量数的相对偏差≤5×10-6,二级碎片离子精确质量数的相对偏差≤10×10-6,保留时间偏移0.5 min,最小响应为10 000,软件匹配度不小于90%。
参考《兽药残留检测中质谱方法确证指南》[24]、《食品法典委员会食品中兽药残留物的最大残留限度(MRLs)和风险管理建议》[25],按照目标分析物的定量下限作为筛查截止浓度进行方法试验,结果表明本筛查方法在截止浓度水平上的假阴性率(β误差)<5%,符合欧盟2002/657/EC决议[26]规定的筛选方法性能指标要求。
2.4 基质效应
通过标准曲线法考察基质效应,分别配制空白基质工作曲线和溶剂标准曲线,按下式计算基质效应(ME):ME=B/A,A和B分别表示溶剂标准溶液和基质标准溶液中药物的峰面积。当ME大于1时,表明存在基质增强效应;当ME小于1时,表明存在基质抑制效应;当ME等于1时,表明无基质效应或基质效应很弱。结果表明,大部分化合物的ME小于1,存在基质抑制效应。因此,本方法采用样品空白基质溶液配制标准曲线进行定量检测。
2.5 方法学验证
根据各药物的检出限选择合适质量浓度范围(0.05~50μg/L)配制基质标准曲线溶液,按本方法进行测定,外标法定量。结果表明,123种药物在各自质量浓度范围内呈良好线性,相关系数(r2)均大于0.99。因目标分析物同时存在禁用和限用药物,所以本方法的定量下限(LOQ)为各目标分析物的最低加标浓度,123种药物的LOQ为0.1~50μg/kg(见表2)。以草鱼空白样品为基质,按各药物的LOQ、2LOQ、10LOQ为加标水平进行回收实验,各加标水平平行测定6次。结果显示,123种药物的回收率为53.7%~129%,相对标准偏差(RSD)为2.7%~19%,方法的准确度和精密度均满足检测要求。
2.6 实际样品检测
采用本方法对100份水产样品(包括草鱼、鲫鱼、黄骨鱼、鲈鱼、多宝鱼等)进行筛查,检测结果与二级谱库的匹配度均大于90%,其中有6个样品检出氯霉素,8个样品检出恩诺沙星,9个样品检出氢化可的松,2个样品检出氧氟沙星。选取4个代表性阳性样品,采用不同检测方法进行确证分析,阳性样品的检测结果见表3和图3。结果表明:阳性结果基本一致,本方法可满足实际检测需要。
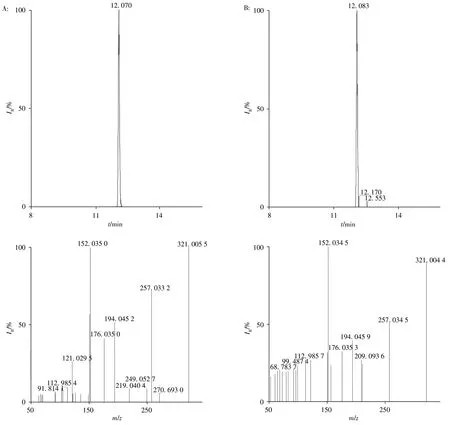
图3 氯霉素标准溶液(A)与多宝鱼阳性样品(B)的谱图Fig.3 Spectra of chloramphenicol standard(A)and a turbot positive sample(B)
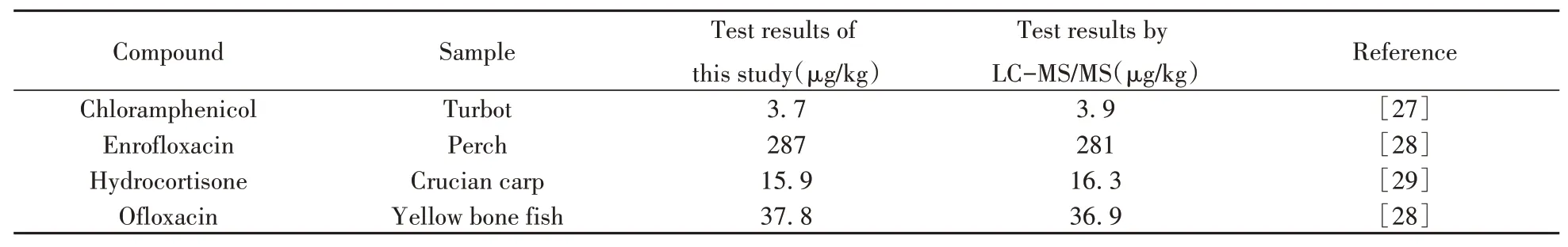
表3 不同方法对阳性样品检测结果的对比Table 3 Comparison of different detection methods for positive samples
3 结 论
本文建立了可高通量快速筛查水产品中123种药物残留的在线净化/液相色谱-四极杆/静电场轨道阱高分辨质谱技术。所建方法的筛查浓度水平能够满足水产品中药物残留管理要求,有效提高了水产品中兽药残留的快速检测水平,对于提高水产品风险监测水平具有重要的意义。
猜你喜欢
当代水产(2022年7期)2022-09-20
煤化工(2022年3期)2022-07-08
小学阅读指南·低年级版(2022年5期)2022-05-09
中山大学学报(自然科学版)(中英文)(2022年2期)2022-04-12
发明与创新·中学生(2018年10期)2018-10-15
食品界(2017年9期)2017-09-30
中国动物保健(2016年3期)2016-05-07
中国信息化·学术版(2013年3期)2013-06-25
