
线粒体融合蛋白 2 与缺血-再灌注损伤
2022-03-17 10:16欧志宇苗芸
器官移植 2022年2期
欧志宇 苗芸
线粒体是细胞中具有重要功能的细胞器,由线粒体外膜(outer mitochondrial membrane,OMM)和线粒体内膜(inner mitochondrial membrane,IMM)组成,两者可动态变化,实现线粒体的运动、融合和分裂[1]。线粒体融合和分裂是线粒体动力学的一部分,对细胞适应内环境变化具有重要意义:能量过剩时,线粒体网络游离化,降低线粒体的能量利用效率,避免能量浪费;能量不足时,线粒体趋向于融合以提高能量利用效率来维持能量供给[2]。其中膜结构的改变在线粒体动力学中具有至关重要的作用。位于OMM上的线粒体融合蛋白(mitofusin,MFN)2是介导线粒体融合的核心蛋白[1]。除介导线粒体融合外,MFN2还可通过调节线粒体自噬、线粒体-内质网结构偶联和细胞凋亡等生理过程参与缺血-再灌注损伤(ischemia-reperfusion injury,IRI)的发生和发展[3]。本文将对MFN2的结构与调节、MFN2的功能及MFN2在IRI中的作用进行综述,以加深对IRI机制的理解,并为IRI防治提供参考。
1 MFN2的结构与调节
MFN是哺乳动物OMM上一类具有三磷酸鸟苷酶(guanosine triphosphatase,GTPase)活性的蛋白,分为MFN1和MFN2两个亚型,二者的同源性约为80%[3]。MFN2由757个氨基酸组成,具有多个结构域,从肽链的N端到C端分别为GTPase结构域、第一个七肽重复(heptad repeat 1,HR1)结构域、脯氨酸富集(proline-rich,PR)结构域、2个相互靠近的跨膜(transmembrane,TM)结构域和第二个七肽重复(heptad repeat 2,HR2)结构域(图1A)[4]。其中,PR结构域只存在于MFN2。由于MFN2和MFN1高度同源,且MFN2包含了MFN1的所有结构域,因此目前的研究主要集中在MFN2上。
MFN2的表达水平会直接影响线粒体融合和分裂之间的平衡,因此受到细胞的严格调控。MFN2的表达主要受过氧化物酶体增殖物激活受体γ共激活因子(peroxisome proliferator-activated receptor gamma coactivator,PGC)-1α 和 PGC-1β 的正向调节[3]。在运动、寒冷等情况下,MFN2主要由PGC-1α诱导以适应高耗能状态,PGC-1β则在平静状态下激活MFN2以维持正常的线粒体稳态[5]。MFN2的降解主要受泛素-蛋白酶体系统影响,如细胞应激情况下,MFN2被c-Jun氨基末端激酶(c-Jun N-terminal kinase,JNK)磷酸化后,会募集泛素连接酶(E3),发生泛素化的MFN2被运送到蛋白酶体中进行降解,引起线粒体断裂和细胞凋亡[6]。
2 MFN2的功能
2.1 介导线粒体融合
线粒体融合是2个线粒体的OMM和IMM在物理结构上合并的过程,融合的线粒体可进行物质交换,有缺陷的线粒体可重新获得呼吸链和线粒体DNA等基本成分,避免了线粒体的损失[7]。OMM融合由MFN介导,MFN聚集在相邻线粒体间的紧密邻近区域,通过HR2结构域形成的同源二聚体(MFN1-MFN1和MFN2-MFN2)或异源二聚体(MFN1-MFN2)连接OMM(图1B),此时GTPase结构域水解三磷酸鸟苷(guanosine triphosphate,GTP)为融合提供能量以完成融合[3-4]。
MFN通过调节线粒体融合影响线粒体的形态。在小鼠胚胎成纤维细胞中敲除MFN1或MFN2都会引起线粒体形态异常,但两者缺陷导致的线粒体形态异常不同,MFN1缺失的线粒体呈现为大小均一的小球体,而MFN2缺失的线粒体则是大小差异很大的球体,其中部分线粒体会过度肿胀,直径超过正常线粒体[8]。在MFN1或MFN2缺陷时,过表达另一个MFN可部分挽救线粒体的融合活性和维持线粒体形态[8],表明MFN1和MFN2在线粒体融合中具有重叠但不完全重合的功能。
2.2 介导线粒体自噬
线粒体自噬是一种线粒体质量控制系统,可在细胞遭受应激时清除受损的线粒体,维持线粒体稳态[9]。线粒体在极端压力下发生损伤时,自噬体选择性地吞噬受损的线粒体,随后转运至溶酶体并与之融合发生降解[10]。MFN2与线粒体自噬密切相关,MFN2缺失会导致自噬体形成减少和自噬体-溶酶体融合缺陷[11],这是线粒体自噬中的两个关键事件。在多种细胞或组织中观察到,敲除MFN2可抑制线粒体自噬并导致大量受损线粒体累积[12-13]。MFN2调节自噬的经典通路是PTEN诱导的推定激酶蛋白1(PTEN-induced putative kinase protein 1,PINK1)-Parkin通路,在线粒体膜电位丧失情况下,PINK1在OMM处聚集并募集Parkin到线粒体将其磷酸化,磷酸化的Parkin可将MFN2泛素化,随后p62作为衔接分子结合泛素化的MFN2和自噬体膜上的微管相关蛋白轻链3(microtubule associated protein light chain 3,LC3),最终进入溶酶体进行降解(图1C)[14]。因此,MFN2介导的线粒体自噬对于维持正常的线粒体功能和细胞稳态至关重要。
2.3 介导线粒体-内质网结构偶联
线粒体-内质网结构偶联又称线粒体相关的内质网膜结构(mitochondria-associated endoplasmic reticulum membrane,MAM),电子显微镜下这两个细胞器之间相互靠近,但不接触,保持着10~40 nm的间隔[15]。它们之间这种特殊的空间结构由细胞器膜上的分子介导,MFN2是其关键的调节因子[16]。与MFN1不同,MFN2还存在于内质网膜上,内质网膜上的MFN2与OMM上的MFN1和MFN2形成异源或同源二聚体,介导MAM的形成[17],敲除MFN2会导致MAM形成减少[12-13,16]。
MAM可调节多种细胞生命活动。脂质合成的关键酶大多位于内质网膜上,由于酶的定位不同,磷脂酰乙醇胺(phosphatidyl ethanolamine,PE)和磷脂酰胆碱(phosphatidylcholine,PC)的合成需依靠线粒体和内质网间进行的脂质运输。位于内质网上的磷脂酰丝氨酸(phosphatidylserine,PS)首先转移到线粒体,然后在IMM中转化为PE,随后转运回内质网并转化为PC[18]。细胞器间的脂质运输对维持线粒体质量有重要意义。研究表明,饥饿诱导的线粒体自噬并不会导致线粒体质量的大幅度下降,其主要原因是线粒体可借助MAM从内质网补充PS和其它磷脂[19]。MAM除了在脂质合成中有重要作用外,也参与调节Ca2+信号传导。MFN2连接线粒体和内质网对Ca2+流动非常重要,Ca2+从内质网转移到线粒体由肌醇1,4,5-三磷酸受体(inositol 1,4,5‐trisphosphate receptor,IP3R)、葡萄糖调节蛋白75(glucose-regulated protein 75,Grp75)和电压依赖性阴离子选择通道蛋白1(voltage-dependent anion-selective channel protein 1,VDAC1)组成的Ca2+通道介导(图1D)[20]。缺乏MFN2会导致细胞器之间的距离增大,无法有效组装Ca2+通道,从而引起线粒体摄取Ca2+减少及Ca2+信号传导的异常。总而言之,MFN2介导的MAM对细胞的基本生命活动调节至关重要。
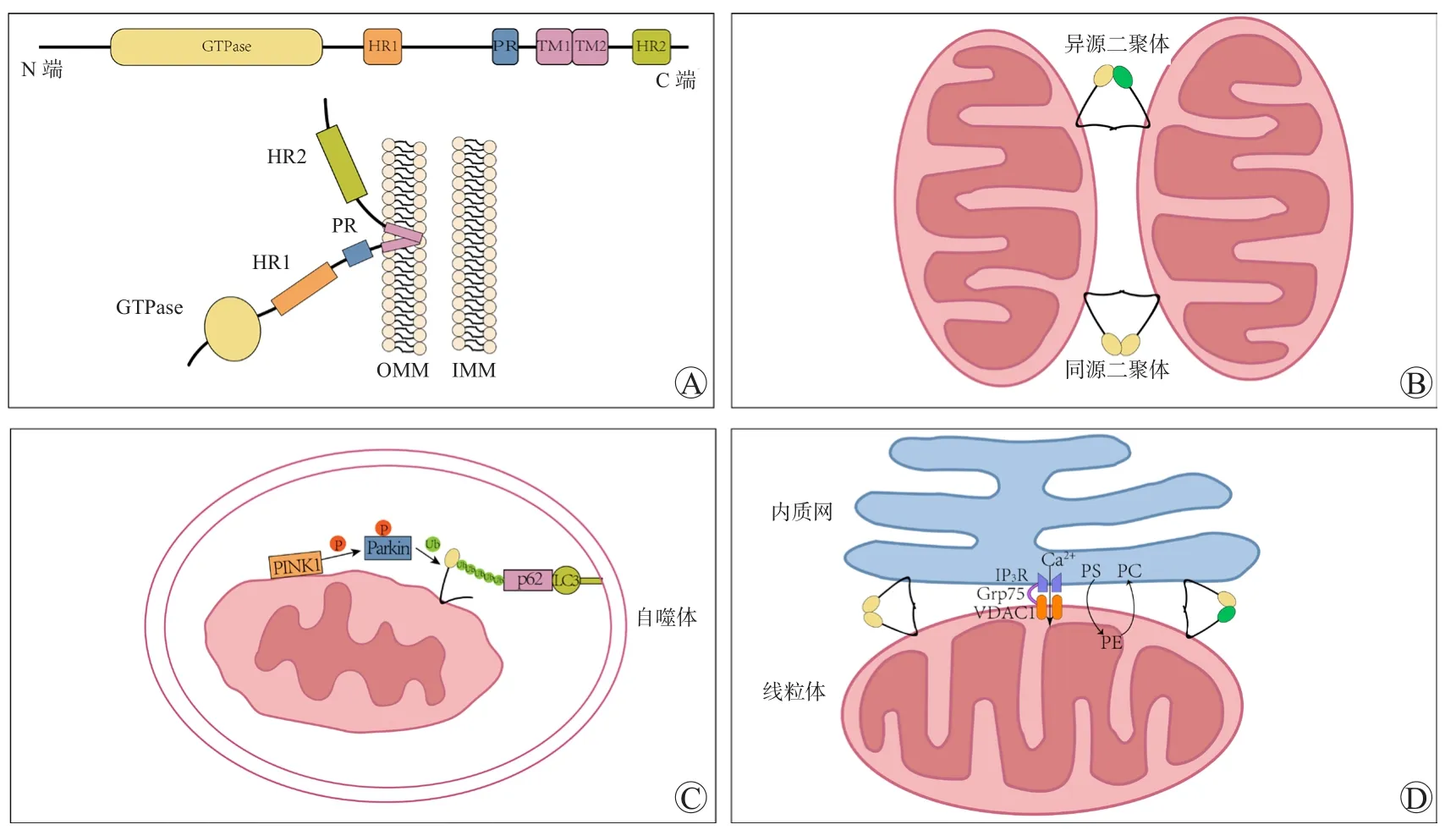
图1 MFN2的结构及其功能Figure 1 Structure and function of MFN2
2.4 调节细胞凋亡
细胞凋亡是一种受调控的细胞死亡过程,通过激活不同的半胱氨酸天冬氨酸蛋白酶(cysteinyl aspartate specific proteinase,Caspase) 启 动 级 联 反应,切割众多底物,最终引起细胞死亡[21]。有证据表明,MFN2能与B淋巴细胞瘤-2相关X蛋白(B cell-lymphoma-2 associated X protein,Bax)、B淋巴细胞瘤-2拮抗剂杀手(B cell-lymphoma-2 antagonist killer,Bak)等凋亡蛋白相互作用以调节细胞凋亡[22]。当线粒体膜电位下降时,MFN2招募Bax到线粒体上,随后Bax和Bak会寡聚化并在OMM形成孔以增加其通透性[23]。线粒体通透性增加后,细胞色素C等促凋亡因子从线粒体膜间隙释放到细胞质中,并与凋亡蛋白酶激活因子1结合形成凋亡体,最终引起细胞凋亡[23]。过表达MFN2可促进Bax从细胞质向线粒体膜转移,从而诱导多种肿瘤细胞凋亡[24-25]。另外,MFN2可通过触发Ca2+从内质网流入线粒体来诱导肝癌细胞凋亡[26]。这些数据都显示了MFN2具有促凋亡作用,然而,也有部分实验证实MFN2具有抗凋亡作用。MFN2泛素化被蛋白酶体降解后会引起线粒体断裂和细胞凋亡增加[6]。敲除MFN2会增强神经元对凋亡刺激的敏感性[27]。MFN2在凋亡中显示出完全相反的作用可能归因于不同的细胞类型、凋亡刺激信号的种类以及病理机制。
3 MFN2与IRI
IRI是导致肾移植术后急性肾损伤和移植物功能延迟恢复的主要原因[28],主要表现为炎症、血液动力学紊乱和内皮细胞损伤。IRI与线粒体损伤、内质网应激、细胞凋亡、氧化应激等事件有关,尤其是线粒体损伤和内质网应激[28]。越来越多的证据表明,MFN2在IRI过程中扮演关键角色。MFN2可通过调节线粒体融合、线粒体自噬、MAM形成等生物过程影响IRI[29-31]。
在IRI过程中,线粒体融合可促进细胞存活。融合使线粒体间物质交换成为可能,使有缺陷的线粒体重新获得呼吸链和线粒体DNA,避免线粒体的损失。Liu等[31]发现神经元细胞IRI后,MFN2的转录和翻译水平都显著下降,并与细胞活力降低和细胞凋亡增多有关。而细胞过表达MFN2时,线粒体融合和膜电位增加,IRI诱导的细胞凋亡受到抑制[31]。有研究报道,抑制MFN2降解和维持MFN2表达可保持心肌细胞线粒体网络的完整性,从而减轻心肌IRI[32]。因此,MFN2可通过促进线粒体融合在IRI中发挥保护作用。
线粒体自噬作为一种选择性自噬,可特异性地清除受损的线粒体,减轻IRI。Peng等[33]研究发现,IRI可激活神经元细胞的自噬,诱导自噬体形成增加,而自噬受损则会加重IRI诱导的神经元损伤。在神经元细胞中过表达MFN2可增加自噬,在脑IRI中发挥保护作用[33]。相反,破坏MFN2介导的线粒体自噬会诱导线粒体损伤从而加重脑IRI[34]。有研究报道,MFN2通过增加自噬在IRI中发挥的保护作用与去乙酰化酶1(sirtuin 1,SIRT1)有关[30]。SIRT1将MFN2去乙酰化可激活自噬,防止IRI后线粒体功能障碍和细胞凋亡,在肝IRI中发挥保护作用[30]。
MAM作为许多反应发生的平台,与众多细胞生命活动相关,如线粒体-内质网间的Ca2+流动、炎症小体激活、脂质代谢、线粒体自噬等。其中MFN2可介导线粒体-内质网间的Ca2+流动,加重IRI。如上所述,在小鼠心脏中特异性敲除MFN1和MFN2可减少MAM形成,从而减少Ca2+从内质网流向线粒体[29],有助于IRI后的细胞存活。同样地,肝IRI模型中观察到,敲除MFN2有助于保留线粒体膜电位和防止线粒体Ca2+超载而发挥保护作用[35]。此外,MAM还可通过引起线粒体Ca2+超载激活NOD样受体蛋白3(NOD-like receptor protein 3,NLRP3)炎症小体[36]。NLRP3炎症小体激活后可通过促进炎症因子释放和介导细胞焦亡来加重IRI。
有趣的是,MAM也能通过介导线粒体自噬在IRI中发挥保护作用。免疫荧光成像数据证实自噬体在MAM处形成[37],自噬体膜可能主要来源于内质网。敲低MFN2会减少MAM的数量,并破坏自噬体的形成[12]。
如上所述,MFN2在IRI中具有多重作用,既可通过介导线粒体融合和线粒体自噬在IRI中发挥保护作用,也可通过介导线粒体-内质网间的Ca2+流动加重IRI。MFN2具体发挥何种作用可能取决于损伤程度以及保护机制和损伤机制之间的平衡:如在轻度IRI情况下,线粒体融合和自噬可清除损伤的线粒体快速恢复细胞稳态,而在严重的IRI状态下,Ca2+超载导致的细胞器损伤难以被线粒体融合和自噬所纠正而表现为细胞死亡。
4 小 结
总而言之,MFN2作为一个具有GTPase活性的OMM蛋白,参与线粒体融合、线粒体自噬、MAM形成和细胞凋亡等生理过程,并通过调节这些生物过程影响IRI进展。尽管众多实验已证实了MFN2对IRI的重要性,但MFN2在IRI中的作用机制尚未完全阐明,需进一步探索,为改善IRI,提高移植物存活率提供理论基础。
猜你喜欢
中国生物化学与分子生物学报(2022年4期)2022-09-07
生物化学与生物物理进展(2022年8期)2022-08-20
湖北农业科学(2022年11期)2022-07-18
临床肝胆病杂志(2021年7期)2021-12-26
心血管病学进展(2021年8期)2021-09-13
现代临床医学(2021年1期)2021-01-26
湖北农业科学(2020年24期)2021-01-21
实用肿瘤学杂志(2020年4期)2020-12-08
癌变·畸变·突变(2015年3期)2015-02-27
癌变·畸变·突变(2015年3期)2015-02-27
