
核磁共振技术在首批中药化学对照品研制中的应用△
2022-03-16 06:07刘静冯玉飞刘阳戴忠马双成
中国现代中药 2022年2期
刘静,冯玉飞,刘阳,戴忠,马双成
中国食品药品检定研究院,北京 100050
核磁共振(nuclear magnetic resonance,NMR)技术是一种非常重要的有机化合物结构解析工具[1-3]。通过解析一维谱(如1H-、13C-NMR)化学位移值、谱峰多重性、偶合常数值、谱峰相对强度和各种二维谱[如异核单量子相关谱(HSQC)、异核多键相关谱(HMBC)、1H-1H COSY]中呈现的相关峰,能够获取定性结构信息。对于结构复杂或结构未知的化合物,通常需要结合其他谱学方法确定其结构。对于结构简单或已知化合物,可直接通过一维谱信号或与文献值比对进行确定。NMR 还可以用于定量分析,以氢核磁共振定量(1H-quantitative nuclear magnetic resonance,1H-qNMR)应用最多,其原理是在合适实验条件下,信号峰面积与产生信号的质子数成正比[4-6]。1H-qNMR 具有样品用量少、测定时间短及无损等优势,目前已广泛应用于药品质量控制等领域,并已收载于《中华人民共和国药典》2020 年版[7]、《美国药典》USP40-NF35[8]和《欧洲药典》10.0版[9]等。
中药化学对照品主要从天然产物中提取精制而得,通常为结构已知成分,但由于天然产物结构复杂多样,常存在异构体,因此,首批对照品的定性鉴别工作仍需高度重视,准确鉴定其结构是对照品准确性的源头保障。在实际工作中,通常采用NMR、质谱、紫外、红外等谱学技术,但NMR 提供的结构信息更为丰富。中药化学对照品的定值目前采用国际通用的质量平衡法,对于首批品种而言,还需要其他定值方法进行佐证,尤以1H-qNMR 应用广泛。本文结合中药化学对照品首批研制实例,阐述了NMR的定性和定量特性,为充分保障首批对照品结构与定值的准确性提供了科学依据。
1 定性鉴定
1.1 结构相近化合物
槲皮素-3-O-β-D-葡萄糖基-(1→2)-α-L-鼠李糖苷(1)和山柰酚-3-O-β-D-葡萄糖基-(1→2)-α-L-鼠李糖苷(2)均属于黄酮苷类化合物,结构相近,两者结构差异在于黄酮苷元B 环(图1)。1H-NMR分析表明,前者B 环为C-3′,4′-二羟基取代,其B环质子H-5′作为1 个二重峰(d,J=8.0 Hz)出现在δ6.70~7.10 处,H-2′(d,J=2.0 Hz)和H-6′(dd,J=8.0,2.0 Hz)信号出现在δ7.20~7.50 处,且通过H-2′和H-6′化学位移可区别黄酮C-3′,4′位上是羟基还是甲氧基;后者B 环为C-4′位羟基取代,其B 环质子分为H-2′,6′和H-3′,5′,构成AA′BB′系统,且H-3′,5′化学位移比H-2′,6′化学位移值小,主要是C-4′位羟基的屏蔽作用及C 环对H-2′,6′的负屏蔽效应。通过13C-NMR 分析,C-2~4 位化学位移表明两者均属于黄酮-3-氧苷类化合物,进一步分析表明其A环为C-5,7-二羟基黄酮类化合物,其C-6和C-8 化学位移信号为δ90.0~100.0,且C-6 信号较C-8 信号处于较低场。此外,对于两者取代糖基的确定,槲皮素-3-O-β-D-葡萄糖基-(1→2)-α-L-鼠李糖苷质谱中出现m/z303 [M+H-162-146]+基峰碎片,山柰酚-3-O-β-D-葡萄糖基-(1→2)-α-L-鼠李糖苷质谱中出现m/z287[M+H-162-146]+基峰碎片,表明两者结构中均存在1 个六碳醛糖和甲基五碳醛糖;结合1H-NMR,通过2个糖端基质子信号偶合常数可判断分别为β-D型(J=7.5 Hz)和α-L型(J=1.0 Hz);进一步综合1H-NMR、13C-NMR 信号确定为β-D-葡萄糖基和α-L-鼠李糖基。通过与文献比对,归属两者核磁信号[10]。槲皮素-3-O-β-D-葡萄糖基-(1→2)-α-L-鼠李糖苷:1H-NMR(600 MHz,CD3OD)δ:7.30(1H,d,J=2.0 Hz,H-2′),7.26(1H,dd,J=8.0,2.0 Hz,H-6′),6.87 (1H,d,J=8.0 Hz,H-5′),6.32(1H,d,J=2.0 Hz,H-8),6.15 (1H,d,J=2.0 Hz,H-6),5.59 (1H,d,J=1.0 Hz,H-1″),4.31 (1H,d,J=7.5 Hz,H-1‴),4.21 (1H,dd,J=3.5,1.5 Hz,H-2″),3.80 (1H,dd,J=11.0,3.5 Hz,H-3″),3.61 (1H,dd,J=12.0,4.5 Hz,H-6‴),3.57 (1H,dd,J=12.5,2.5 Hz,H-6‴),0.92 (3H,d,J=6.0 Hz,H-6″);13C-NMR (125 MHz,CD3OD)δ:159.3 (C-2),136.5 (C-3),179.6 (C-4),163.2 (C-5),99.9 (C-6),166.0 (C-7),94.7 (C-8),158.5 (C-9),105.9 (C-10),122.8 (C-1′),116.9 (C-2′),146.5 (C-3′),149.9 (C-4′),116.4 (C-5′),122.8(C-6′);鼠李糖基:102.6 (C-1″),82.9 (C-2″),71.7(C-3″),73.5 (C-4″),72.0 (C-5″),17.7 (C-6″);葡萄糖基:107.2 (C-1‴),75.3 (C-2‴),77.8 (C-3‴),70.7(C-4‴),77.8(C-5‴),62.1(C-6‴)。山柰酚-3-O-β-D-葡萄糖基-(1→2)-α-L-鼠李糖苷:1H-NMR (600 MHz,CD3OD)δ:7.72 (2H,dd,J=7.0,2.0 Hz,H-2′,6′),6.89(2H,dd,J=7.0,2.0 Hz,H-3′,5′),6.32(1H,d,J=2.0 Hz,H-8),6.15 (1H,d,J=2.5 Hz,H-6),5.67 (1H,d,J=1.0 Hz,H-1″),4.36 (1H,d,J=7.5 Hz,H-1‴),4.23(1H,dd,J=3.5,1.5 Hz,H-2″),3.74 (1H,dd,J=10.0,3.5 Hz,H-3″),3.66 (1H,dd,J=12.0,2.5 Hz,H-6‴),3.61 (1H,dd,J=12.0,4.5 Hz,H-6‴),0.88(3H,d,J=7.5 Hz,H-6″);13C-NMR (125 MHz,CD3OD)δ:159.4 (C-2),136.4 (C-3),179.5 (C-4),163.2 (C-5),100.0 (C-6),166.1 (C-7),94.8 (C-8),158.6 (C-9),105.9 (C-10),122.5 (C-1′),132.0 (C-2′),116.6 (C-3′),161.7 (C-4′),116.6 (C-5′),132.0(C-6′);鼠李糖基:102.5 (C-1″),82.7 (C-2″),71.7(C-3″),73.4 (C-4″),72.0 (C-5″),17.6 (C-6″);葡萄糖基:107.1 (C-1‴),75.3 (C-2‴),77.8 (C-3‴),70.9(C-4‴),77.9(C-5‴),62.3(C-6‴)。
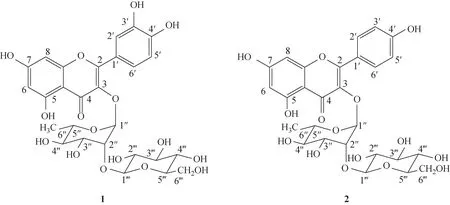
图1 槲皮素-3-O-β-D-葡萄糖基-(1→2)-α-L-鼠李糖苷(1)和山柰酚-3-O-β-D-葡萄糖基-(1→2)-α-L-鼠李糖苷(2)的化学结构式
1.2 同分异构体
地黄苷D(3)为环烯醚萜苷类成分,与文献报道的苦郎树苷C(sammangaoside C,4)[11]为同分异构体(图2),两者主要结构差异在于C-5位糖基连接位置不同。结合文献报道[11-13],通过1H-NMR、13C-NMR 数据分析初步推断为地黄苷D;进一步通过HSQC、HMBC 等二维谱图分析,特别是HMBC 谱中葡萄糖端基质子δ4.92 (H-1″)与苷元C-5 位信号δ84.0 相关,葡萄糖端基质子信号δ4.82 (H-1‴)与碳信号δ82.8(C-2″)相关,以及葡萄糖基质子信号δ3.64(H-2″)与葡萄糖端基碳信号δ105.8(C-1‴)相关,由此确定环烯醚萜苷元C-5 位2 个葡萄糖基为1→2连接,即证明本品为地黄苷D,而非苦郎树苷C,同时明确地黄苷D信号归属:1H-NMR(600 MHz,D2O)δ:5.41
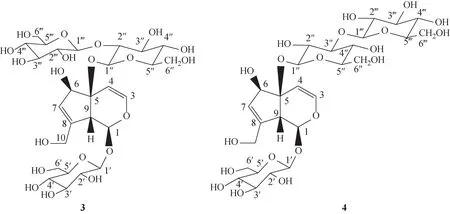
图2 地黄苷D(3)和苦郎树苷C(4)的化学结构式
(1H,d,J=5.4 Hz,H-1),6.55 (1H,d,J=6.6 Hz,H-3),5.28 (1H,d,J=6.6 Hz,H-4),4.52 (1H,s,H-6),5.87(1H,dd,J=1.8,1.2 Hz,H-7),3.31(1H,m,H-9),4.30(1H,d,J=15.6 Hz,H-10),4.24 (1H,d,J=15.6 Hz,H-10),4.78 (1H,d,J=7.8 Hz,H-1′),4.92 (1H,d,J=7.8 Hz,H-1″),4.82(1H,d,J=7.8 Hz,H-1‴);13C-NMR(150 MHz,D2O)δ:98.6 (C-1),146.6 (C-3),107.0 (C-4),84.0 (C-5),83.0 (C-6),130.5 (C-7),147.4 (C-8),54.1 (C-9),62.5 (C-10),101.0 (C-1′),75.7 (C-2′),78.3 (C-3′),72.3 (C-4′),79.0 (C-5′),63.4 (C-6′),99.2 (C-1″),82.8 (C-2″),78.4 (C-3″),72.2 (C-4″),78.8 (C-5″),63.3 (C-6″),105.8 (C-1‴),76.8 (C-2‴),78.5(C-3‴),72.5(C-4‴),79.2(C-5‴),63.7(C-6‴)。
1.3 立体异构体
甲基莲心碱(5)为双苄基异喹啉类生物碱成分,与蝙蝠葛碱(6)为立体异构体(图3),两者主要结构差异在于C-1′位立体构型不同。在结构鉴定过程中,通过解析1H-NMR、13C-NMR谱图,并与文献比对[14],初步鉴定为甲基莲心碱。对于其结构中2 个手性中心立体结构的确定方法,经文献调研可知,甲基莲心碱及其立体异构体蝙蝠葛碱中C-1和C-1′位的立体构型,可根据C-9、C-9′及C-4、C-4′的化学位移差值来确定[15-16]。通过本品核磁碳谱数据解析结果可知,C-9、C-9′及C-4、C-4′的化学位移差值分别为Δδ0.8、Δδ0.9,符合文献报道Δδ(C-9/C-9′)0.7,Δδ(C-4/C-4′)0.9,因此确定本品C-1 和C-1′位立体构型分别为R、S型,即为甲基莲心碱,信号归属为:1H-NMR (600 MHz,CDCl3)δ:3.59 (1H,dd,J=6.6,6.0 Hz,H-1),2.73,3.13 (各1H,m,H-3),2.53,2.82 (各1H,m,H-4),6.50 (1H,s,H-5),6.62 (1H,s,H-8),2.67(1H,m,H-9),3.04(1H,dd,J=5.4,13.8 Hz,H-9),6.90 (2H,d,J=8.4 Hz,H-11,15),6.69 (2H,d,J=8.4 Hz,H-12,14),2.49 (3H,s,C2-NCH3),3.72(3H,s,C6-OCH3),3.54 (3H,s,C13-OCH3),3.63 (1H,dd,J=6.0,6.0 Hz,H-1′),2.73,3.13 (各1H,m,H-3′),2.60,2.82 (各1H,m,H-4′),6.01 (1H,s,H-5′),6.38 (1H,s,H-8′),2.78 (1H,m,H-9′),2.99 (1H,dd,J=5.4,13.8 Hz,H-9′),6.55 (1H,d,J=1.8 Hz,H-11′),6.84 (1H,d,J=8.4 Hz,H-14′),6.69 (1H,重叠,H-15′),2.45 (3H,s,C2′-NCH3),3.79 (3H,s,C6′-OCH3),3.80 (3H,s,C7′-OCH3);13C-NMR (150 MHz,CDCl3)δ:64.4 (C-1),46.7 (C-3),25.2 (C-4),129.2(C-4a),111.1 (C-5),147.1 (C-6),148.9 (C-7),112.3(C-8),131.1 (C-8a),40.6 (C-9),131.9 (C-10),130.4(C-11),113.4 (C-12),144.7 (C-13),113.4 (C-14),130.4 (C-15),42.7 (C2-NCH3),55.4 (C6-OCH3),55.1(C13-OCH3),64.7 (C-1′),47.2 (C-3′),26.1 (C-4′),125.7 (C-4a′),110.9 (C-5′),145.4 (C-6′),146.3 (C-7′),119.2 (C-8′),130.5 (C-8a′),39.8 (C-9′),131.4(C-10′),120.0(C-11′),142.8(C-12′),157.7(C-13′),115.4 (C-14′),125.2 (C-15′),42.5 (C2′-NCH3′),55.8(C6′-OCH3′),55.7(C7′-OCH3′)。
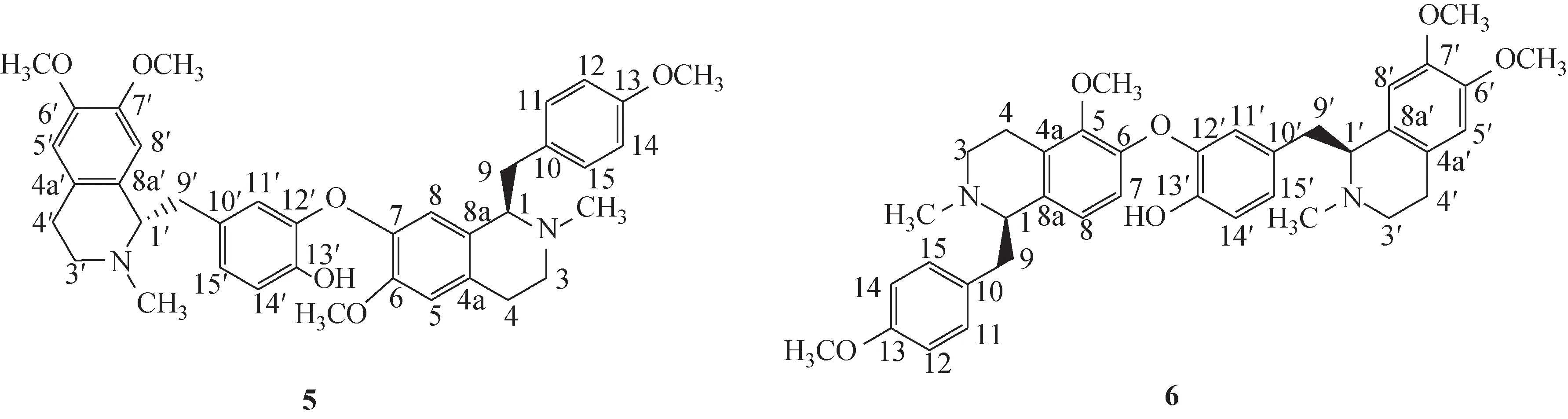
图3 甲基莲心碱(5)和蝙蝠葛碱(6)的化学结构式
25S-鲁斯可皂苷元(7)及其立体异构体25R-鲁斯可皂苷元(8)均属螺甾烷衍生物,分属螺甾烷醇和异螺甾烷醇类成分,其差别在于两者C-25位构型不同,分别为S构型和R构型(图4)。文献报道两者可通过13C-NMR谱中C-23~27位化学位移值可加以区分,其中25S-鲁斯可皂苷元相应化学位移值分别为δ26.5(C-23)、26.3 (C-24)、27.6 (C-25)、65.2 (C-26)、16.4(C-27);25R-鲁斯可皂苷元相应化学位移值分别为δ32.1(C-23)、29.5(C-24)、30.7(C-25)、67.1(C-26)、17.4(C-27)[17-18],在实际工作中,可参照此规律用于确定鲁斯可皂苷元C-25位立体构型。
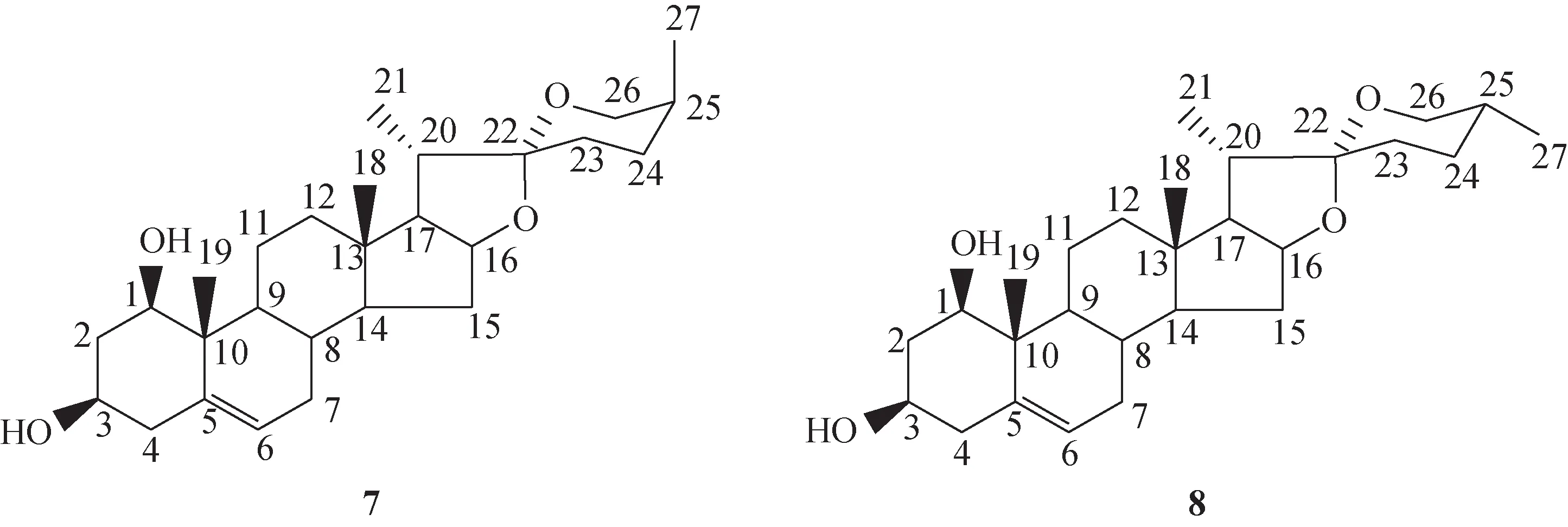
图4 25S-鲁斯可皂苷元(7)和25R-鲁斯可皂苷元(8)化学结构式
2 定量分析
1H-qNMR 应用于首批中药化学对照品辅助定值时常采用内标法,按照公式(1)计算化合物含量。
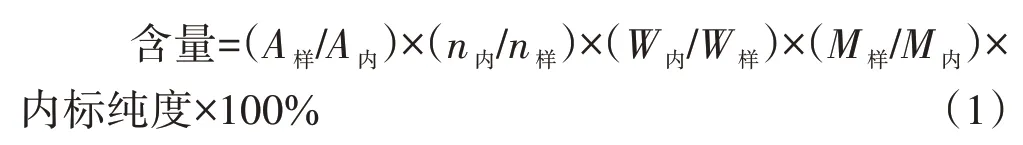
其中,A样为样品选定信号的积分面积;A内为内标选定信号的积分面积;n样为样品被积分信号包含的质子数;n内为内标被积分信号包含的质子数;W样为样品称样量;W内为内标称样量;M样为样品相对分子质量;M内为内标相对分子质量[19]。
2.1 含残留溶剂对照品
1,4-二[4-(葡萄糖氧)苄基]-2-异丁基苹果酸酯质量平衡法定值结果为95.61%。为更好地佐证定值结果的准确性,平行制备3 份样品进行1H-qNMR分析,以马来酸作为内标,氘代甲醇为溶剂,以样品中δ7.07 处氢信号为定量峰,δ6.32 氢信号为内标峰(图5),根据公式(1)计算结果为97.32%,RSD 为0.41%,与质量平衡法结果基本一致。需要特别指出的是,本品IH-NMR、I3C-NMR 谱图定性分析结果表明存在乙酸乙酯的相关信号,后经气相色谱法(GC)验证,外标法确定本品残留溶剂为乙酸乙酯,外标法计算其质量分数为1.73%,RSD 为1.28%;在1H-qNMR 分析时,以乙酸乙酯δ4.10 氢信号作为定量峰,计算其质量分数为1.67%,RSD为3.41%,与GC 测定结果基本一致。由此可见,1H-qNMR 可同时一步测定供试品及其所含残留溶剂含量,结果可靠,操作简便快速。
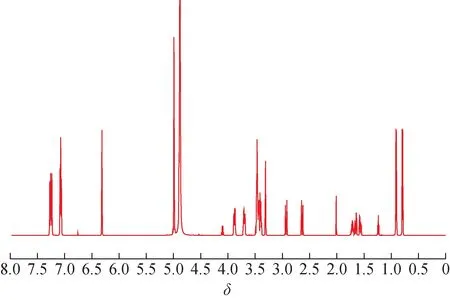
图5 1,4-二[4(-葡萄糖氧)苄基]-2-异丁基苹果酸酯的1H-qNMR谱图
2.2 其他对照品
地黄苷D 质量平衡法定值结果为95.55%。在进行1H-qNMR 分析时,以邻苯二甲酸氢钾作为内标,氘代水为溶剂,以样品中δ5.87 处氢信号为定量峰,邻苯二甲酸氢钾δ7.73 氢信号为内标峰,根据公式(1)计算结果为94.21%,RSD 为0.80%,与质量平衡法结果基本一致。
甲基莲心碱质量平衡法定值结果为99.09%。在进行1H-qNMR 分析时,以对苯二甲酸二甲酯作为内标,氘代三氯甲烷为溶剂,以样品中δ6.39 处氢信号为定量峰。对苯二甲酸二甲酯δ8.10 氢信号为内标峰,根据公式(1)计算结果为99.53%,RSD 为0.58%,与质量平衡法结果基本一致。
需要说明的是,尽管氢核磁共振定量法较质量平衡法操作简便,但测定结果的准确性也受一些因素影响,包括溶剂选择、内标物选择、溶解度及仪器相关参数设置等,在实际操作过程中应结合具体品种加以摸索优化[19]。
3 结论
中药化学对照品的重要性不言而喻,准确鉴定结构及确定其含量是保障发放品种准确的重要源头环节。中药化学对照品尽管是已知化合物,但由于其结构类型复杂多样,常存在结构相近、同分异构体及立体异构体等情况,NMR 可以提供化学位移值、偶合常数、谱峰多重性等丰富信息,能够揭示化合物的结构片段信息甚或立体结构,在中药化学对照品结构鉴定工作中具有不可替代的作用。此外,1H-qNMR 需要样品量少、无损、快速,可一步测定供试品含量及残留溶剂等有关物质,是目前常用的中药化学对照品首批品种定值佐证方法。
猜你喜欢
口腔护理用品工业(2021年4期)2021-11-02
昆明医科大学学报(2021年8期)2021-08-13
保健与生活(2021年4期)2021-02-22
家庭百事通·健康一点通(2020年11期)2020-11-30
中国科技纵横(2019年23期)2019-02-14
军营文化天地(2018年2期)2018-04-20
小学生时代·大嘴英语(2017年1期)2017-03-20
海峡科技与产业(2017年1期)2017-03-04
爆笑show(2015年9期)2015-10-24
中国信息化·学术版(2013年3期)2013-06-25
