
以兰索拉唑肠溶胶囊为例探索模型引导的仿制药等效性替代方法*
2022-03-11 09:23张锦琳宋芸峰贾欢欢袁耀佐
药学与临床研究 2022年1期
张锦琳,李 岩,陈 涛,周 颖,3,宋芸峰,3,贾欢欢,4,袁耀佐**,张 玫,曹 玲
1 江苏省食品药品监督检验研究院,国家药品监督管理局化学药品杂质谱研究重点实验室,南京 210019;2上海凡默谷信息技术有限公司,上海 200127;3南京中医药大学,南京 210023;4中国药科大学,南京 211121
兰索拉唑是日本武田公司研制的质子泵抑制药,其抑制胃酸分泌作用及抗幽门螺旋杆菌作用较强,在治疗消化性溃疡疾病方面有着显著疗效。但因其对酸不稳定,口服时多制成肠溶制剂。天津武田和日本武田的兰索拉唑肠溶胶囊均为国家药品监督管理局公布的参比制剂,国内还有较多兰索拉唑肠溶胶囊的仿制药,但因肠溶胶囊辅料多、工艺复杂,仿制药与参比制剂往往形似而神不似,按质量标准检验均合格,但临床疗效差异较大,即现有质量标准无法保证产品的疗效,因此药品监管和制药企业迫切需要建立一种通过体外检验反映体内疗效的质量控制方法。本研究将探索建立体内外相关性与生物等效性的替代方法。
本研究通过搭建兰索拉唑的体内PK 模型,反推口服给药后在胃肠道内的释放与吸收曲线,进而构建兰索拉唑肠溶胶囊的体内外相关性模型,模拟出生物体相关的溶出方法。联合仿制药的体外溶出曲线研究结果,进而实现在不进行临床BE 研究的情况下,仅通过比较该方法汇总的仿制药与参比制剂体外溶出曲线,即可虚拟评估仿制药与参比制剂的等效性情况。
1 软件和材料
1.1 软件
PBPK 建模软件GastroPlus(version 9.0.0003,Simulations Plus,Inc.,USA);Digit(version 1.0.4,Simulations Plus,Inc.,USA);体外崩解与溶出模拟软件DDDPlus(version 5.0.0011,Simulations Plus,Inc.,USA)。
1.2 药品
兰索拉唑肠溶胶囊(规格:30 mg),分别来自天津武田药品有限公司和7 家仿制药生产企业(企业A~G)。
2 方法与结果
2.1 验证性PK 模型的搭建
收集兰索拉唑建模所需的各种理化与生物药剂学性质参数,如溶解度、渗透性、脂溶性、血浆蛋白结合率等[1-4],主要模型参数见表1。
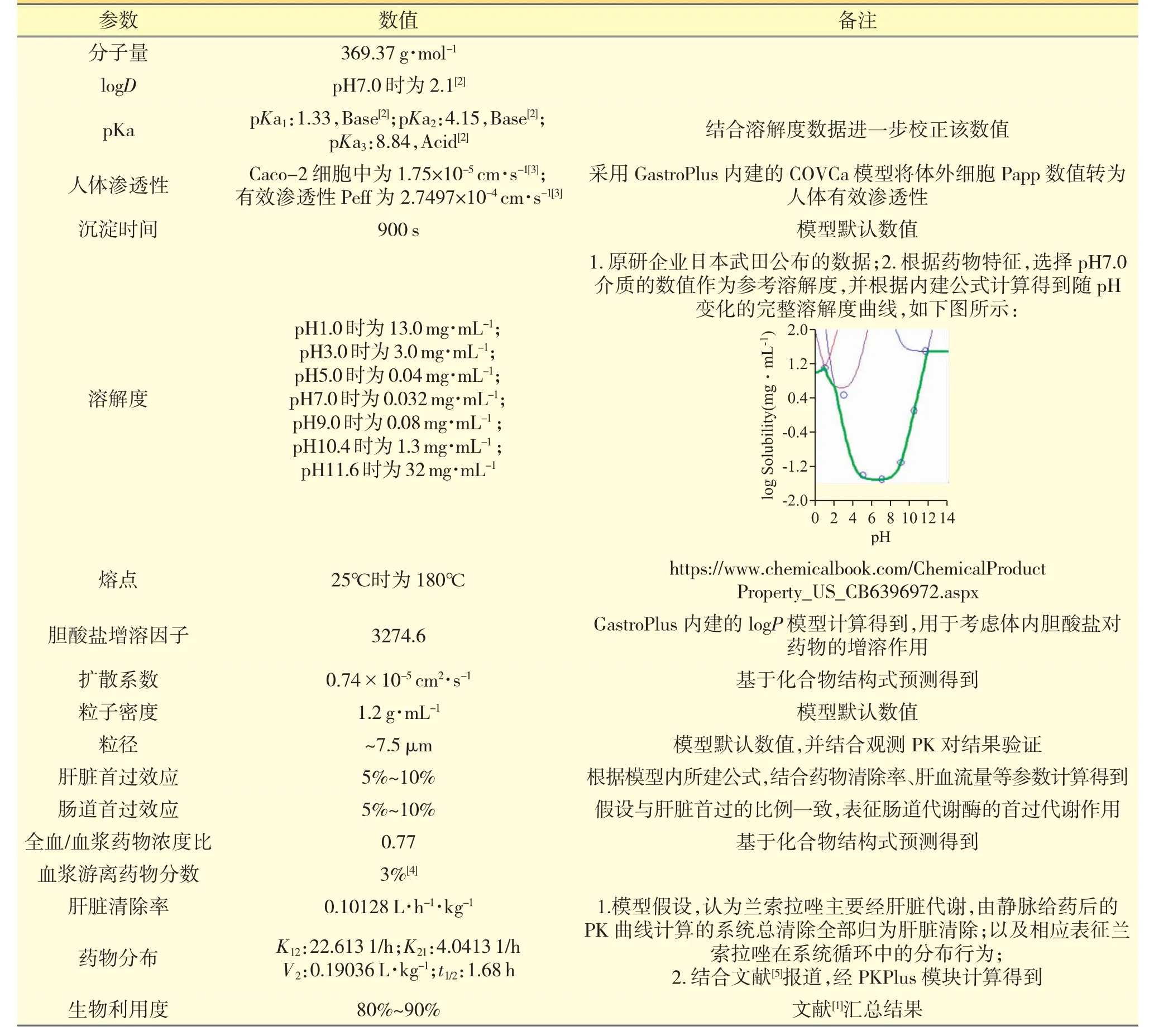
表1 兰索拉唑PK 模型建立过程中的主要模型参数
主要来自文献、软件内建模以及基于化合物结构式预测的结果。查阅、提取文献报道的兰索拉唑经静脉或口服给药后在人体的血药浓度-时间数据点;整理归纳所收集的数据,并综合文献报道的信息,全面梳理模型药物的体内吸收、分布、代谢、排泄特点,以辅助更加准确地搭建药物PK 模型,验证PK 模型搭建的相关参数见表2。

表2 兰索拉唑肠溶胶囊验证PK 模型相关参数
以文献报道的口服给药[6]后的PK 数据作为参照,通过GastroPlus 软件搭建兰索拉唑的吸收和处置模型,并设置与文献报道中相同的给药途径、给药剂量、受试者人群等相关信息,然后进行血浆PK曲线的预测,强代谢人群和弱代谢人群口服参比制剂后PK 模拟结果分别见图1 和图2,并比对预测与实测曲线间的吻合程度(拟合度R2)、主要PK 参数(Cmax,Tmax,AUC)的预测值与观测值的偏差(相关结果见表3),以确认所建立的模型是否可以重现已有的临床结果,从而构建兰索拉唑可靠、稳健的GastroPlus PK 模型[7,8]。以全面考察及验证Gastro-Plus 模型对兰索拉唑人体内的吸收、代谢和分布过程的拟合准确性,确定建模过程中的所有ADME 性质参数,以及相应的模型计算公式等。
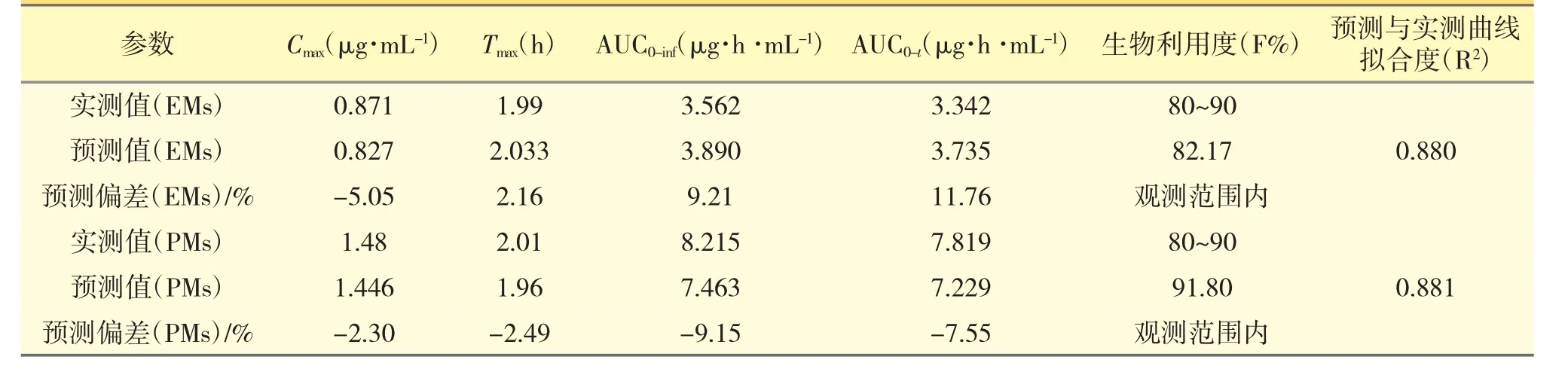
表3 EMs 与PMs 人群口服参比制剂后,观测数据与预测偏差信息
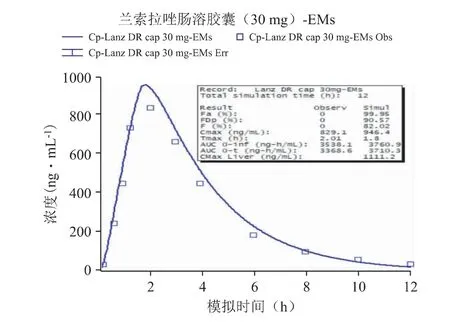
图1 EMs 口服参比制剂后PK 模拟结果
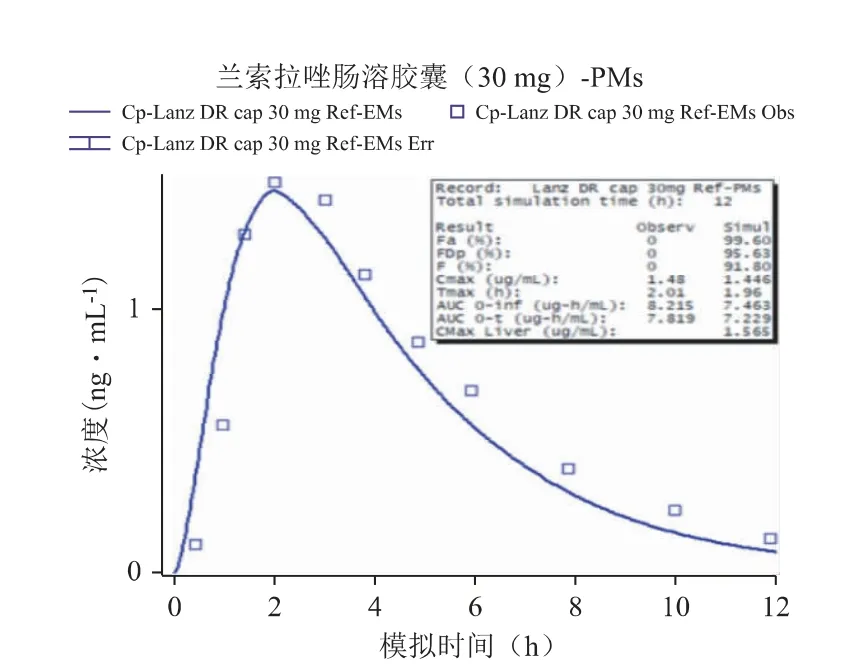
图2 PMs 口服参比制剂后PK 模拟结果
2.2 兰索拉唑法定溶出条件体内外相关性评价
在“2.1”验证后的兰索拉唑体内PK 模型基础上,借助机制性口服吸收模型(GastroPlus 的ACAT模型)反推得到兰索拉唑(30 mg 肠溶胶囊)在胃肠道中的释放与吸收曲线,见图3;30 mg 兰索拉唑肠溶胶囊肠道的局部吸收特征见图4。
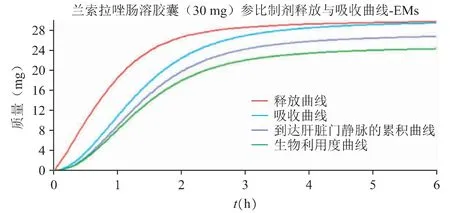
图3 30mg 兰索拉唑肠溶胶囊在胃肠道中的释放与吸收曲线
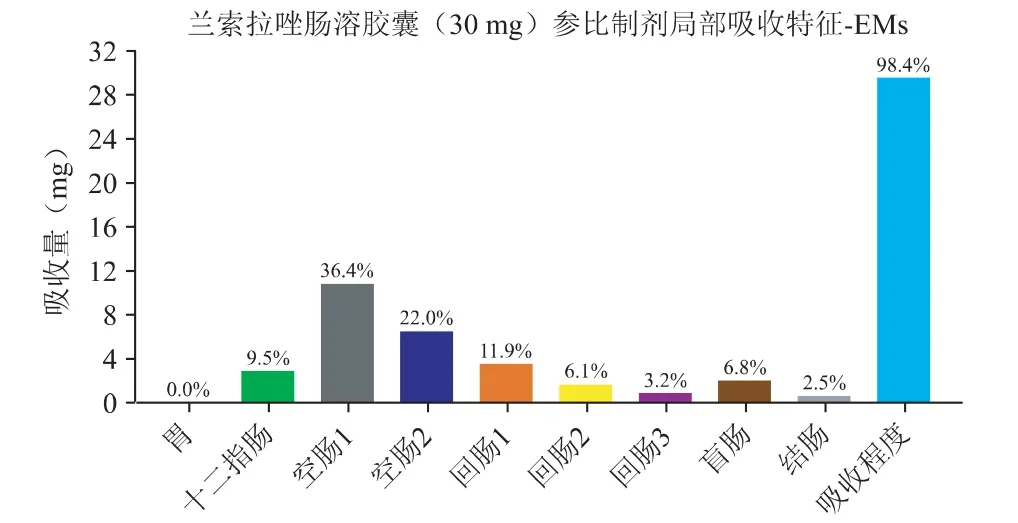
图4 30 mg 兰索拉唑肠溶胶囊胃肠道的局部吸收特征
口服给药后兰索拉唑在胃肠道中基本可以完全跨膜吸收,主要的吸收部位是十二指肠和空肠。图3 中可以看出,由于兰索拉唑在吸收部位(pH 5~7)的溶解度较差,使得口服肠溶胶囊后在体内呈现缓慢释放的行为,约2 个小时达到完全释放;浅蓝色的吸收曲线稍滞后于体内释放曲线,溶出为分子状态的药物可以快速吸收进入肠细胞,并在溶出完全时完成吸收过程。预估体外溶出与体内释放之间可以建立一定的相关性,限制肠溶胶囊制剂吸收快慢的主要因素为药物的释放速率。
本研究分别采用pH 6.8 和pH 6.0 两种溶出介质考察参比制剂的体内外相关性:将各介质的溶出数据点加载至模型,然后通过Weibull 方程,将体外各时间点的累积溶出度拟合为体内连续溶出曲线,通过比对基于不同溶出介质曲线预测得到的PK曲线,与文献中或临床实测PK 曲线之间的拟合程度,分析制剂体外溶出曲线与体内PK 曲线之间的相关特征[9,10],Weibull 方程如下:
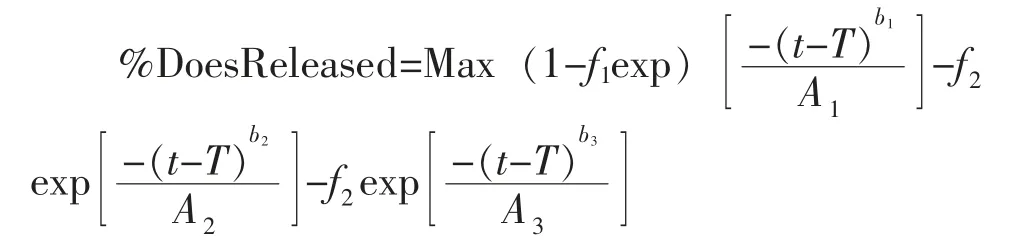
其中,Max 为总溶出量(%);T 为溶出滞后时间(h);f1为1 相的溶出分数,f2为2 相的溶出分数,f3为3 相的溶出分数;b1、b2和b3分别为1 相、2 相和3相的形状因子;A1、A2和A3分别为对应相的时间放大因子。
采用Weibull 方程处理兰索拉唑参比制剂于pH 6.8 介质下的溶出曲线,示意图见图5。代入上述两个介质测定的溶出曲线至肠溶胶囊参比制剂30 mg 的模型中,并利用体外溶出数据预测对应的体内PK 曲线,发现预测结果与该制剂临床报道的血药浓度-数据点均有一定的差异:这两个介质预测的PK 曲线均低于观测结果,曲线的吻合度R2仅在0.6 左右(其中pH 6.8 的吻合度稍好于pH 6.0 的结果),Cmax分别约低10%和25%,AUC 的预测与观测结果较为一致,Tmax出现较大的差异(均快于观测值25%以上,这是导致预测曲线拟合度较差的主要原因),说明直接采用这两个介质的溶出曲线不能很好地预测体内PK 变化(见图6、图7)。

图5 pH 6.8 介质下溶出曲线的Weibull 方程(图中数据点为体外实测的溶出数值;线为根据实测数据拟合的Weibull释放曲线;左侧列举了对应的Weibull 方程参数)
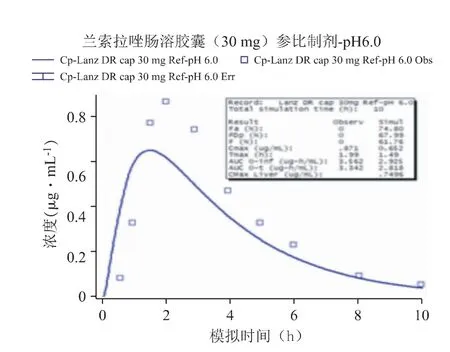
图6 代入体外溶出曲线(pH 6.0)预测的PK 曲线
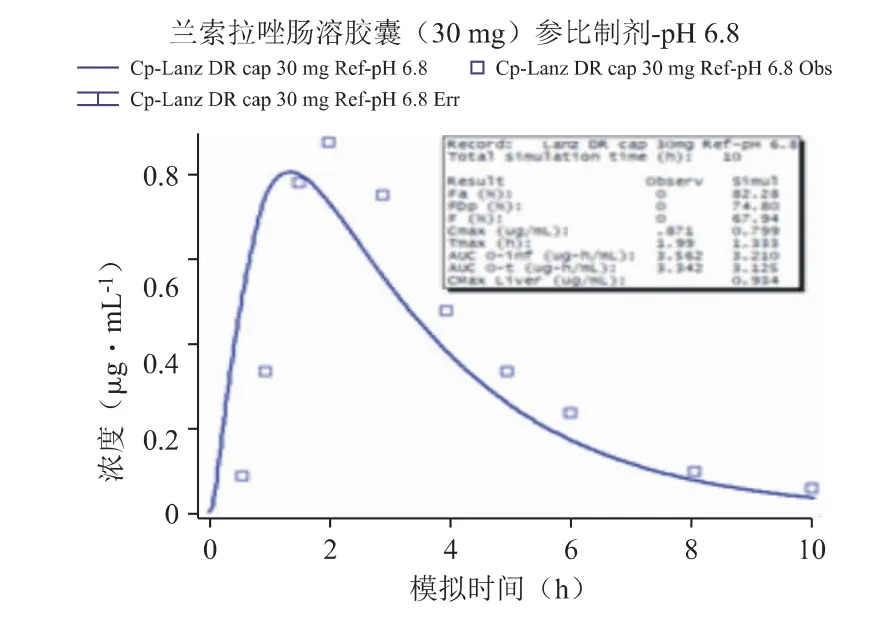
图7 代入体外溶出曲线(pH 6.8)预测的PK 曲线
当前研究还进一步使用GastroPlus 软件的体内外相关性(In Vivo In Vitro Correlations,IVIVC)模块,将体外与体内累积释放百分数进行相关性分析,结果见图8、图9。实线为相同时间点体外与体内释放点所作的图,虚线为基于相关性方程拟合的曲线。图8 结果表明,pH 6.0 的曲线建立的相关系数R2较低,不能很好地进行体内外转化,特别是累积释放较高时呈现出较大的误差。pH 6.8 溶出介质中建立的曲线相关系数较高,且总体曲线的重合度较好,可以较好地反映药物在体内的释放行为。因此,综合Weibull 方程的PK 曲线预测结果以及IVIVC 的研究分析,可以发现体外pH 6.8 条件下的溶出行为与体内释放具有一定的相关性,趋势一致,但不是生物体相关的溶出方法,即每个时间点的体外释放量完全对应体内释放的量。

图8 pH 6.0 体外溶出与体内累积释放量之间的相关性
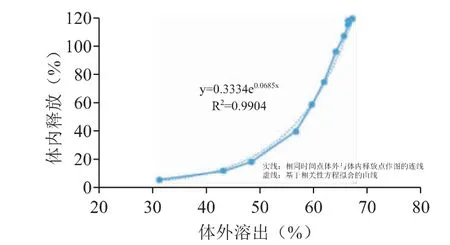
图9 参比制剂在pH 6.8 溶出介质体外溶出与体内累积释放量之间的相关性
由于当前的体外溶出曲线与体内的释放过程有一定的差异,无法真实反映该药物在体内的释放的每一个时间点特性。借助体外崩解与溶出模拟软件DDDPlus 考察体外溶出行为,通过该溶出模型,可结合肠溶胶囊的处方、溶出方法与条件等参数,进行相应溶出曲线的模拟;同时以参比制剂体内释放曲线为目标,逐步优化体外溶出条件,使得模型预测的溶出曲线与目标体内释放曲线一致,如图10 所示,以辅助开发出生物体相关的溶出方法。通过调整溶出条件使得预测的溶出曲线和体内释放点吻合一致,以建立反映体内释放过程的生物体相关性溶出方法。模型最终推测的生物体相关性溶出方法为:采用USP I 法的篮法;设定仪器转速为100 r·min-1;并在45 min 之前采用pH 5.5 的磷酸盐缓冲液、45 min之后调整溶出介质的pH,形成pH 为7.4 的缓冲液作为溶出实验的介质;介质体积为900 mL。
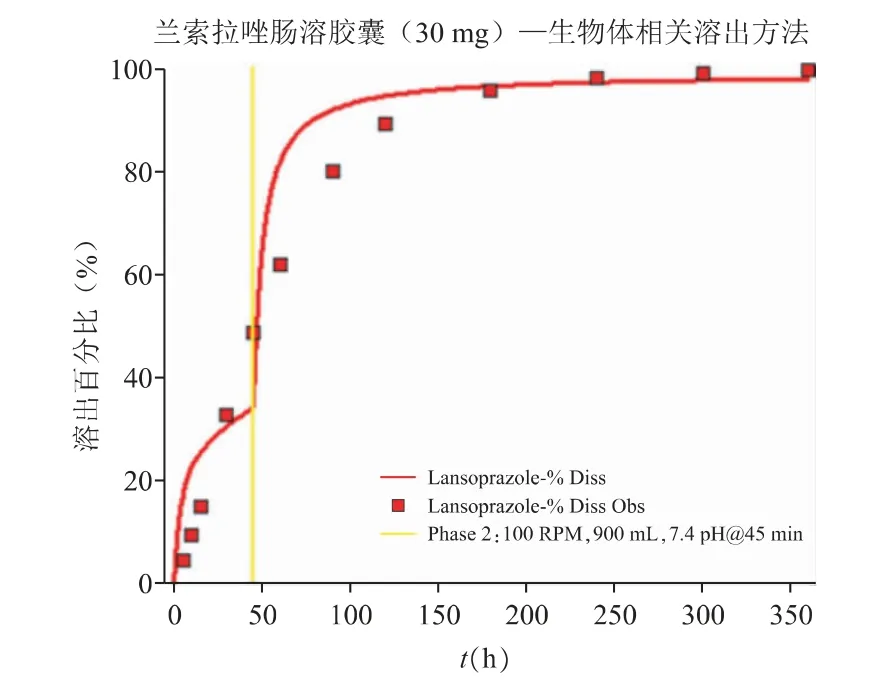
图10 生物体相关溶出方法的优化结果
2.3 兰索拉唑肠溶胶囊仿制制剂与参比制剂虚拟生物等效性评估
基于pH 6.8 介质下测定的溶出曲线和体内有一定的相关性,采用该溶出条件测定各制剂的体外溶出曲线,基于相关性的转换方程以得到对应的体内释放数据点,然后借助GastroPlus 软件的Weibull方程转化为体内连续释放曲线。
由图11 体外数据结果显示,A、B、C、D 4 家企业的仿制药呈现快速及完全地释放行为,在当前溶出条件下基本在30 min 内即可溶出完全,尤其C、D 2 家的释放速率较参比制剂更为迅速。E、F、G 3 家仿制药则呈现较为缓慢且不完全的释放行为,其中企业E 的初始释放速率与参比制剂最为相似,但是超过2 h 时,该制剂表现为一定程度的降解,导致末端的释放与参比制剂有一定的差异;企业F 及企业G 的制剂虽然初始释放行为稍快于参比制剂,但最终的释放程度较为接近参比制剂。
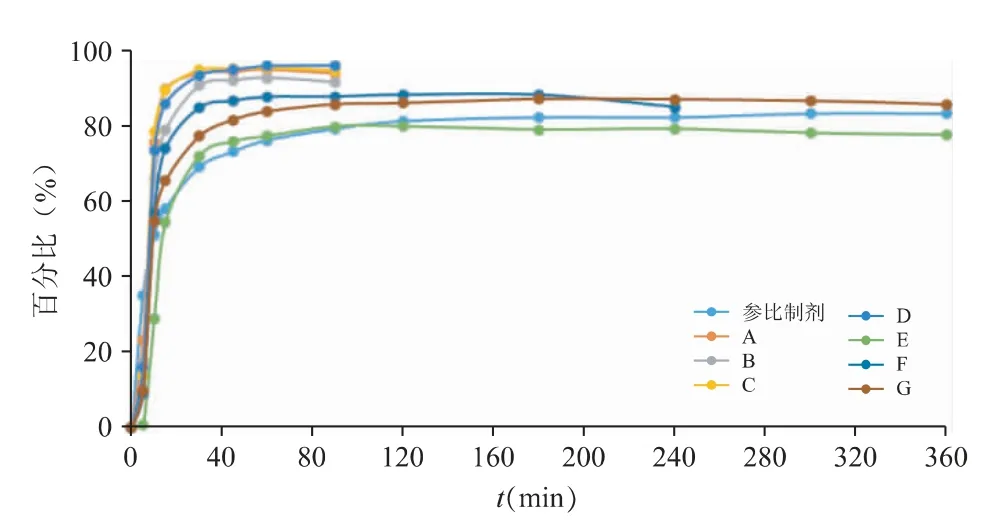
图11 pH 6.8 介质下各仿制制剂体外溶出曲线
将各制剂的体外溶出数据用软件转化为图12体内释放曲线,转化之后的曲线缩小了各仿制药与参比制剂间的溶出差异。除企业E 外,其它6 家制剂均可在一定时间内达到体内完全释放,而参比制剂完全释放则需要3~4 h,这可能使这些厂家的制剂预测的体内达峰时间Tmax要小于参比制剂的结果,同时也可能由于快速释放引起Cmax的不等效;而企业E 的释放速度虽然最为接近参比制剂的特征,但是末端的不完全释放,则可能使Cmax降低,造成Cmax和AUC 的不等效。

图12 各仿制制剂经相关性转化的体内释放曲线
借助GastroPlus 的群体PK 模拟以及虚拟BE评估功能,GastroPlus 的群体模拟功能一次可虚拟产生2500 个受试者,并通过蒙特卡洛抽样法[11]产生虚拟人群数据,包括胃肠道生理参数、PK 参数、制剂参数、给药方案和化合物理化性质参数等,对上市仿制药与参比制剂执行交叉虚拟等效性试验。进行参比与受试制剂的群体PK 模型的设置,并虚拟48 个受试者进行PK 曲线的预测以及生物等效性的统计分析。模型分别评估了7 个仿制药与参比制剂的等效性情况,发现来自企业A、B、F 及G 的仿制药可能与参比制剂生物等效;而来自企业C、D 及E 的仿制药则与参比制剂的主要PK 参数(Cmax,AUC)的统计学结果落在等效性范围外,提示可能与参比制剂生物不等效。
现仅对企业A 和企业C 作出分析:企业A 与参比制剂的虚拟生物等效性评估结果见图13;企业C 与参比制剂的虚拟生物等效性评估结果见图14。
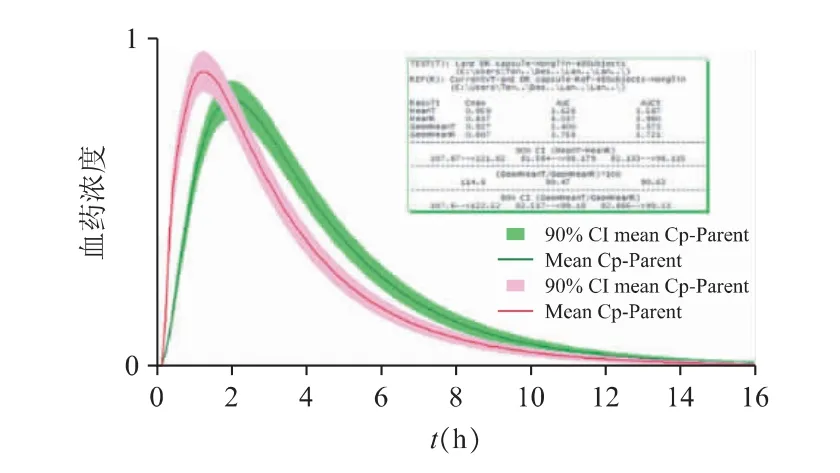
图13 企业A 与参比制剂虚拟生物等效性评估结果(绿色为参比制剂,红色为仿制制剂)
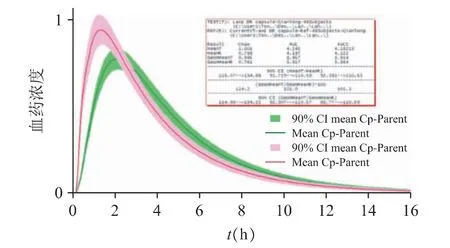
图14 企业C 与参比制剂虚拟生物等效性评估结果(绿色为参比制剂,红色为仿制制剂)
企业A 的仿制药Cmax与AUC 均较接近参比制剂的结果,对数转换后的Cmax的90%置信区间为107.60%~122.52%、对数转换后的AUC0-inf的90%置信区间为82.52%~99.18%,提示可能与参比制剂生物等效。但该制剂的体外溶出数据(pH 6.8)显示,初始的释放行为要明显快于参比制剂,可能会导致Tmax的差异,并进而引起Cmax的不等效,需要结合参比制剂的肠溶包衣特点,进一步优化肠溶包衣工艺;或优化当前的体外溶出方法。
企业C 仿制药的Cmax高于参比制剂的结果,但两者的AUC 较为接近。经对数转换后的Cmax的90%置信区间为114.93%~134.22%、对数转换后的AUC0-inf的90%置信区间为92.31%~110.57%,提示可能与天津武田的参比制剂生物不等效。其可能的原因是企业C 的仿制药在体外表现出较快的释放行为,进而加快了血浆中药物浓度的达峰,造成Cmax超出了等效性的上限。
各仿制药的体内释放要快于参比制剂,导致仿制药的Tmax较短、PK 曲线的行为有一定差异,此外较快的释放速度,也容易引起峰浓度Cmax的不等效,因此联合生物体相关的溶出方法,深入考察各仿制药与参比制剂的释放差异,并确定等效的可能性是较为合适的方式。
3 讨论
随着仿制药质量与疗效一致性评价的进程,仅仅用体外溶出曲线评价仿制药与参比制剂的质量差异,而没有进一步关联制剂体内过程,忽略体内外相关性模型的搭建,具有一定的局限性。从国外研究情况来看,美国、日本、欧盟、加拿大等多国的药监部门早已引入软件,以辅助进行药物体内行为的模拟、制剂体内外相关性的建立、生物等效性的研究等,提高了药品审评审批的进度与合理性。通过软件的预测,可模拟药物在体内的行为,从而结合评价药品在体内特征,来制定体外溶出度标准,建立体外溶出曲线与体内药代曲线的相关性。可大大减少体外试验的数量与成本,同时能获得更合理及有区分力的体外溶出方法,真实反映仿制药的内在质量。PBPK 模型在仿制药的研发中潜力很大,但在应用中也需进行体内外数据的验证,充分理解该制剂的吸收机理[12],考虑临床研究数据的平行性与可比性。目前,在应用上PBPK 模型还存在一些挑战,如个体血药浓度-时间数据点较难获得、部分品种静脉给药的PK 数据查询不到、体内外相关性溶出方法较难建立等[12]。
在模拟出可能生物体相关的溶出条件后,后续研究将在此条件下,以溶出曲线数据和收集到该品种的BE 数据,进行比对验证,进一步来证实该替代方法的可行性和可靠性,为药物的质量控制及一致性评价提供一种简单、便捷的手段、为科学监管提供新的思路。
猜你喜欢
现代仪器与医疗(2022年3期)2022-08-12
社会科学战线(2022年3期)2022-06-15
现代装饰(2021年2期)2021-07-21
医学前沿(2021年18期)2021-04-14
今日农药(2017年2期)2017-03-24
山东工业技术(2017年1期)2017-01-24
今日农药(2016年6期)2016-05-14
小学阅读指南·高年级版(2009年3期)2009-03-27
家庭医药(2009年1期)2009-02-05
中学生数理化·高二版(2008年4期)2008-11-12
