
Toxicities of amyloid-beta and tau protein are reciprocally enhanced in the Drosophila model
2022-03-09 07:33:42ZhenDongSunJiaXinHuJiaRuiWuBingZhouYunPengHuang
中国神经再生研究(英文版) 2022年10期
Zhen-Dong Sun, Jia-Xin Hu, Jia-Rui Wu, Bing Zhou, Yun-Peng Huang
Abstract Extracellular aggregation of amyloid-beta (Aβ) and intracellular tau tangles are two major pathogenic hallmarks and critical factors of Alzheimer’s disease. A linear interaction between Aβ and tau protein has been characterized in several models. Aβ induces tau hyperphosphorylation through a complex mechanism;however, the master regulators involved in this linear process are still unclear. In our study with Drosophila melanogaster, we found that Aβ regulated tau hyperphosphorylation and toxicity by activating c-Jun N-terminal kinase. Importantly, Aβ toxicity was dependent on tau hyperphosphorylation, and flies with hypophosphorylated tau were insulated against Aβ-induced toxicity. Strikingly, tau accumulation reciprocally interfered with Aβ degradation and correlated with the reduction in mRNA expression of genes encoding Aβ-degrading enzymes, including dNep1, dNep3, dMmp2, dNep4, and dIDE. Our results indicate that Aβ and tau protein work synergistically to further accelerate Alzheimer’s disease progression and may be considered as a combined target for future development of Alzheimer’s disease therapeutics.
Key Words: Alzheimer’s disease; amyloid-beta; amyloid-beta degradation; Drosophila melanogaster; c-Jun N-terminal kinase (JNK); neurodegeneration; tau; tau hyperphosphorylation
Introduction
Alzheimer’s disease (AD) is the most common type of human dementia and causes memory loss, intellectual impairment, cognitive decline,language disability, and several other social disabilities. AD is predominantly categorized as sporadic AD, and only 5% of AD cases are caused by inherited mutations in proteins, such as amyloid beta precursor protein (APP),presenilin 1, and presenilin 2. Several risk factors have been suggested,including apolipoprotein E ɛ4 polymorphism, which is strongly correlated with AD pathology (Yamazaki et al., 2019), imbalance of the gut microbiota(Goyal et al., 2021), dyshomeostasis of trace metal elements (Lei et al.,2021), over-activation of microglia (Schwabe et al., 2020), and impaired exosomes (Zhang et al., 2021). Although some risk factors of sporadic AD have been characterized, the cause of sporadic AD remains unknown. Besides apolipoprotein E ɛ4, some genetic loci, such as phosphatidylinositol binding clathrin assembly protein and clusterin, are involved. Additionally, potential causes of AD include metabolic-related factors, such as hypercholesterolemia,type 2 diabetes, and hyperhomocysteinemia and inflammatory cues, such as neuro-inflammation and abnormal microglial activation (Ballatore et al.,2007; Chakrabarti et al., 2015). Characterization of core risk factors and axes for sporadic AD is paramount at present and will aid the further exploration of the mechanisms of sporadic AD.
Although the mechanism of AD is debated, AD can be pathologically characterized by the extracellular aggregation of amyloid beta (Aβ) plaques and intracellular tau tangles (Foidl et al., 2020; Mamun et al., 2020). Aβ is a short peptide composed of 39-42 amino acids that is generated and released from the amyloidogenic processing of APP by beta- and gamma-secretases(Chen et al., 2017). Aβ is the major component of senile plaques and is believed to be a cause of neuronal damage in the brains of AD patients (Chen et al., 2017; Zeng et al., 2021). Toxicity of Aβ has been investigated in multiple models (Chen et al., 2000; Newman et al., 2010; Prüβing et al., 2013).However, increasing evidence suggests that the microtubule-associated protein tau acts as the central player in AD pathology rather than Aβ (Bloom,2014). Tau was initially characterized as a microtubule-associated protein that binds to and stabilizes microtubules. Under pathological conditions, such as AD and frontotemporal lobar degeneration, tau is hyperphosphorylated by several kinases, including microtubule affinity regulating kinase 2 (MARK2)PAR-1, FYN, GSK3, and CDK5. Hyperphosphorylation of tau decreases microtubule binding affinity and axon localization, after which tau is released into the neuronal cell body and post-synaptic compartments. Subsequently,hyperphosphorylated tau forms oligomeric aggregates and tangles, which can further block the function of neurons and cause deleterious effects (Hoover et al., 2010; Srivastava et al., 2021).
According to the linear model of Aβ and tau interaction, Aβ disrupts neuronal function and induces tau dysregulation, and tau then acts as a downstream effector to execute Aβ toxicity; down-regulation of endogenous tau can effectively block Aβ toxicity (Bloom, 2014). Although some mediators, such as the FYN kinase and NMDA receptor, are suggested to be involved in the linear interaction process (Ittner et al., 2010; Bloom, 2014), the mechanism and the core mediators are still unclear.
Drosophila melanogaster(D. melanogaster) is widely used in studies of human neurodegenerative diseases (Bilen and Bonini, 2005). The majority of fundamental biological pathways are conserved between the fly and human genomes according to comparative genomics studies, and nearly 70% of human disease-related genes have orthologs in the fly genome (Fortini et al.,2000). Compared with other model organisms,D. melanogasteroffers many advantages, such as a short lifespan, an abundance of genetic tools, and easy access for phenotypic characterization. These advantages can benefit the analyses of fundamental molecular and pathological mechanisms in complex human diseases, including human neurodegenerative diseases (Bilen and Bonini, 2005).
At present, various human neurodegenerative diseases can be modeled inD. melanogaster, including AD, Parkinson’s disease, Huntington’s disease,and amyotrophic lateral sclerosis. Expression of human Aβ42or tau in the fly central nervous system (CNS) can be used to investigate some of the pathological features of human AD, such as cognitive decline, behavioral dysfunction, pathological aggregate formation, reduction of synaptic transmission and neuronal mitochondrial homeostasis, and shortened lifespan(Link, 2005; Prüβing et al., 2013). Indeed, theD. melanogasterAD model has been widely used to dissect the fundamental mechanisms of AD (Wittmann et al., 2001; Shulman and Feany, 2003; Folwell et al., 2010; Sowade and Jahn, 2017; Jeon et al., 2020). In this study, we used theDrosophilamodel to further investigate the relationship between Aβ and tau. Our results suggest that Aβ and tau can reciprocally aggravate their toxicities to promote AD progression.
Materials and Methods
Fly stocks and crosses
The Aβ fly line used in this study was UAS-Aβ(Arc2E) (designated as Aβ*)and was obtained from Tsinghua University (Beijing, China). The tau fly line was UAS-Tau(R406W) (designated as Tau*). The tau mutant line Tau(S2A)was UAS-Tau(R406W, S262A, S356A) (designated as Tau*S2A). The UASTau(R406W) and UAS-Tau(R406W, S2A) flies were obtained from Stanford University (Palo Alto, CA, USA). The wild-type control, GMR-Gal4, and ELAVGal4 flies were previously purchased from the BloomingtonDrosophilaStock Center (Indiana University; Bloomington, IL, USA). The GMR-Gal4Sm6a;UAS-Tau(R406W)Tm6B flies and GMR-Gal4; UAS-Tau(R406W, S2A)Sm6a fly crosses were generated as previously described (Huang et al., 2015). Similarly,ELAV-Gal4 flies were crossed with UAS-Tau(R406W) and UAS-Tau(R406W,S2A) flies to obtain ELAV-Gal4Y; UAS-Tau(R406W)Tm6B and ELAV-Gal4Y;UAS-Tau(R406W, S2A)Sm6a flies. These flies were then crossed with UASAβ(Arc2E) flies to obtain GMR-Gal4UAS-Aβ(Arc2E); UAS-Tau(R406W)+, GMRGal4, UAS-Tau(R406W, S2A)UAS-Aβ(Arc2E), ELAV-Gal4+; UAS-Aβ(Arc2E)+;UAS-Tau(R406W)+, and ELAV-Gal4+; UAS-Aβ(Arc2E)UAS-Tau(R406W, S2A)flies. The GMR-Gal4UAS-GFP and ELAV-Gal4+; UAS-GFP+ flies were used as controls (Table 1). Flies were maintained on a standard corn-based medium at 25°C. The c-Jun kinase (JNK) inhibitor SP600125 (Cat# S5567, MilliporeSigma,Burlington, MA, USA) was added to the medium at a final concentration of 10 μM. Vitamin C (Cat# 50-81-7, Sinopharm, Shanghai, China) was added to the medium at a final concentration of 5 mM to reduce the level of reactive oxygen species (ROS).

Table 1 |Fly information
Observations of Drosophila eye phenotypes and scanning electron microscopy (scanning-EM)
The rough eye phenotype was observed and quantified according to severity(Crowther et al., 2005; Oh et al., 2015). Representative photographs were captured using an XTL-165-MT microscope (Phenix; Jiangxi, China). The rough eye phenotype was divided into four groups for quantification as follows: severe: +++, medium: ++, slight: +, and normal: -. The numbers of corresponding rough eye phenotypes were counted and analyzed. For the scanning-EM observations, adult flies were quickly frozen in liquid nitrogen and adhered to the scanning-EM objective table. Images of fly eyes were observed using the FEI Quanta 2000 Scanning Electron Microscope (FEI Company, Hillsboro, OR, USA) as described previously (Huang et al., 2014)under the following conditions: voltage, 15 kV; pressure, 300 Pa; and 4°C with 36.9% humidity.
Climbing ability assay and lifespan analyses
To determine climbing ability (Madabattula et al., 2015), newly eclosed female flies were collected and divided into three to six individual groups, and each group included 20 adult flies. Flies were aged at 25°C for 3-4 weeks, and the numbers of flies that climbed 3 cm in 8 seconds were manually counted and divided by the total number of flies, which was then used as the climbing index (%). To record fly lifespans, newly eclosed flies were collected and maintained at 25°C, fly food was exchanged every 2 days, and the numbers of dead flies were counted. For each genotype, at least 80-100 newly eclosed flies were used to record lifespans.
Separation of soluble and insoluble protein fractions and western blots
To separate the soluble and insoluble tau fractions, 50 adult fly heads were collected, homogenized in NP40 protein extraction buffer (Beyotime; Cat#P0013; Shanghai, China), and centrifuged at 12,000 ×gfor 10 minutes.The supernatant was centrifuged again at 100,000 ×gfor 30 minutes and then collected as the soluble fraction. The pellet was resuspended in NP40 protein extraction buffer containing 1% SDS (Cat# A100227, Sangon Biotech,Shanghai, China) and collected as the insoluble fraction. Protein samples were mixed with 5× SDS-loading buffer, denatured at 37°C for 30 minutes,and loaded on 12% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) gels(Cat# B546013, Sangon Biotech) to separate proteins. The separated proteins were transferred on to a 0.22 μm polyvinylidene fluoride membrane (Cat#ISEQ00010, Millipore, Shanghai, China). The membrane was hybridized for 12 hours with mouse anti-tau antibody (clone Tau-5; 1:1500 dilution; Innovative Research Cat# AHB0042, RRID: AB_1502093, Thermo Fisher Scientific;Waltham, MA, USA) and rabbit anti-tubulin antibody (1:2000 dilution; Cat#ab6046, RRID: AB_2210370, Abcam, Cambridge, MA, USA) and rinsed threetimes with 1× phosphate-buffered saline containing 0.1% Tween 20. The blot was then hybridized with the corresponding secondary antibodies for 2 hours(horseradish peroxidase [HRP]-conjugated rabbit anti-mouse IgG, 1:2000,Cat# 7076S, RRID: AB_330924 and HRP-conjugated goat anti-rabbit IgG,1:2000, Cat# 7074S, RRID: AB_2099233; both from Cell Signaling Technology;Shanghai, China). The immunoblot signals were observed using enhanced chemiluminescence (Cat# 34075, Thermo Fisher Scientific). Western blot results were analyzed and quantified using ImageJ software (v1.8.0, National Institutes of Health, Bethesda, MD, USA; Schneider et al., 2012), and the levels of soluble and insoluble tau were normalized to β-tubulin levels.
To separate soluble and insoluble Aβ fractions, 50 adult fly heads were collected. The soluble fraction was collected using the NP40 protein extraction buffer after centrifugation at 100,000 ×gfor 30 minutes as described above.To dissolve the Aβ aggregates and collect the insoluble fraction, NP40 extraction buffer was mixed with formic acid to obtain a 70% formic acid extraction buffer, which was used to dissolve the pellets. The samples were sonicated for 2 minutes to break up Aβ aggregates using a JY92-llDN sonicator(HUXI Company, Shanghai, China). Soluble and insoluble Aβ fractions were mixed with 5× SDS-loading buffer and loaded onto 15% SDS-PAGE gels.Western blotting was performed as described above using rabbit anti-Aβ antibody (1:1000, Cat# 14974P, RRID: AB_2798671, Cell Signaling Technology),mouse anti-β-actin (1:2000, Cat# TA-09, RRID: AB_2636897, ZSGB-Bio, Beijing,China), and the corresponding secondary antibodies (HRP-conjugated rabbit anti-mouse IgG and goat anti-rabbit IgG as described above). Western blots were analyzed and quantified as described above, and the levels of soluble and insoluble Aβ were normalized to β-actin levels.
To detect tau phosphorylation and JNK activation, at least 25 adult fly heads were collected and homogenized in NP40 protein extraction buffer containing proteinase and phosphatase inhibitors. After centrifuging for ten minutes at 12,000 ×g, the supernatant was mixed with 5× SDS-loading buffer and denatured. The proteins were separated and blotted as described above.The signal for phosphorylated (p)-JNK was normalized to total JNK. The phosphorylated tau signals (pS262, PHF-1, and AT8) were normalized to total tau signals using ImageJ software. The following antibodies were used:mouse anti-AT8 antibody (1:1500 dilution; a gift from Stanford University, Palo Alto, CA, USA); mouse anti-PHF-1 antibody (1:2000 dilution; Developmental Studies Hybridoma Bank; University of Iowa; Iowa City, IA, USA); rabbit anti-pS262 (1:1000; Cat# 44-750G, RRID: AB_2533743, Thermo Fisher Scientific),rabbit anti-JNK (1:1500 dilution, Cat# sc-571, RRID: AB_632385, Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-p-JNK (rabbit, 1:2000, Cat#4668P, RRID: AB_823588; Cell Signaling Technology). For these western blot experiments, the primary antibodies were incubated for 12 hours at 4°C; the secondary antibodies were incubated for 2 hours at room temperature.
To analyze tau phosphorylation in mammalian cells, a pcDNA3.1 plasmid construct with Tau(R406W)-RFP sequences (designated as Tau*-RFP) was transfected into HEK293 cells (obtained from the National Collection of Authenticated Cell Culture; Shanghai, China) to generate cell lines with Tau*-RFP expression. The pcDNA3.1 vector alone was transfected to generate a control cell line. Cells were treated with 10 μM Aβ40peptide for 24 hours and harvested for western blot analysis. The levels of tau phosphorylation and p-JNK were analyzed by western blot as described above, and the signals were normalized to Tau5 or JNK respectively, using ImageJ software.
RNA extraction and gel-based reverse transcription PCR analysis
Gel-based reverse transcription PCR (RT-PCR) (Mitchell and Iadarola, 2010)was used to analyze the mRNA expression level of Aβ-degrading enzymes(Additional Table 1). For fly RNA extraction, 30 adult fly heads were collected and homogenized in Trizol buffer (Thermo Fisher Scientific). The cDNA was synthesized using a reverse transcription kit (Cat# AE311; TransGen Biotech;Beijing, China). For the calculation and quantification of RT-PCR results, RTPCR samples were separated on 1.5% agarose gels (Cat# A620014, Sangon Biotech), and the intensity of the DNA bands was evaluated using ImageJ software and normalized to the intensity of the band for ribosomal protein 49 (rp49), which was used as the loading control (Cerro-Herreros et al.,2017). Primer sequences used for RT-PCR analysis ofdNep1,dNep2,dNep3,dIDE, andrp49were previously described (Lang et al., 2013), and the primer sequences fordBace,dMmp1,dMmp2,dNep4,dNepl21,dNepl7, anddAncewere obtained from theDrosophilaprimer Bank (Hu et al., 2013) (DRSC Fly Primer Bank: https://www.flyrnai.org/flyprimerbank). The primer sequences are listed in Table 2.

Table 2 |Primer sequences used for reverse transcription PCR
Statistical analysis
Biochemical analyses were blinded to fly genotypes and treatments, whereas the fly phenotyping was not blinded to the experimental conditions. The data are presented as the mean ± SEM. Differences among groups were analyzed by GraphPad Prism, version 8.0.1 for Windows (GraphPad Software, Inc., San Diego, CA, USA). Unpairedt-tests were used for comparisons of two groups,and one-way ANOVA was used for comparison of multiple groups and to compare the means of pre-selected pairs of columns.P< 0.05 was considered statistically significant.
Results
Aβ expression aggravates tau protein toxicity in the Drosophila model
To investigate the influence of Aβ on tau protein toxicity in the fly model,we co-expressed UAS-Aβ(Arc2E), a more toxic mutant of Aβ, and UASTau(R406W) (designated as Aβ* and Tau*, respectively) inDrosophilacompound eyes by using the GMR-Gal4 driver (Additional Figure 1A, model preparation). Tau* expression caused the rough eye phenotype because of its toxic effect on photoreceptor neurons, and Aβ* co-expression further aggravated this phenotype. We classified the rough eye phenotypes into four groups according to severity. Tau* expression alone caused medium rough eyes, whereas Aβ* expression alone did not result in a rough eye phenotype at 25°C. The co-expression of Aβ* and Tau* increased the ratio of severe rough eyes from approximately 30% to 80% (Figure 1A and B). These results suggested that Aβ aggravated tau protein toxicity. To confirm this result,we investigated the fly eye phenotype using the scanning-EM microscope and found that the rough eye phenotype caused by Tau* expression was exacerbated by co-expression of Aβ* (Figure 1C). The severity of the fusion of ommatidia, dents in the retina, and reduced eye size of Tau* flies was exacerbated by Aβ* expression (Prüβing et al., 2013).
Tau expression not only causes rough eyes inD. melanogaster, it also shortens the lifespan and reduces climbing ability (Gistelinck et al., 2012). Therefore,we evaluated the lifespan and climbing ability of Tau* and Aβ*Tau* flies by using the ELAV-Gal4 driver (Additional Figure 1B, model preparation).Aβ* expression further reduced the medium lifespan of Tau* flies (Figure 1D); the medium lifespan (50% survival) for Tau* flies was reduced from approximately 37 to 25 days after co-expression of Aβ* (Figure 1D). As a control, the medium lifespan for Aβ* flies was approximately 33 days (Figure 1D). The climbing ability assay also showed similar trends. Aβ* co-expression significantly reduced the climbing ability of Tau* flies; the climbing index was reduced from 60% for Tau* flies compared with 8% for Aβ*Tau* flies (Figure 1E). As a control, the climbing index for Aβ*-expressing flies was 40%. The above results suggested that tau protein toxicity was significantly aggravated by Aβ.
Aβ aggravates tau protein hyperphosphorylation and accumulation
Because hyperphosphorylation is the major driving force for tau polymerization and toxicity, we analyzed tau phosphorylation using phosphorylation-specific antibodies, especially to detect the AD pathologyrelated phosphorylated epitopes, such as Ser396Ser404 (PHF-1 epitope),Ser202Thr205 (AT8 epitope), and Ser262. Typically, Ser262 is one of the regulatory phosphorylation sites of tau protein, which mediates subsequent tau hyperphosphorylation on PHF-1(Ser396, Ser404) and Ser202 (Rissman et al., 2004; Nishimura et al., 2004).
Our western blot results demonstrated that Aβ* expression significantly increased the level of tau hyperphosphorylation in Tau* flies; phosphorylation levels on Ser262, PHF-1, and AT8 epitopes all increased when Aβ* was coexpressed with Tau* (Figure 2A and B). Ser262 and PHF-1 signals were also significantly increased in Tau*-RFP cells treated with Aβ40peptide (Figure 2C and D), indicating Aβ can significantly increase tau hyperphosphorylation.We determined the total levels of tau protein in Aβ*Tau* flies through the separation and analysis of the soluble and insoluble tau fractions. Aβ* coexpression significantly increased the level of soluble and insoluble tau protein (Figure 2E and F), indicating that Aβ can exacerbate tau accumulation and hyperphosphorylation as well as toxicity.
Tau hyperphosphorylation mediates the toxicity of Aβ
To further confirm the effect of Aβ on tau hyperphosphorylation, we used Tau*S2A flies that expressed a hypophosphorylated tau mutant. Tau*S2A flies carry alanine substitutions on both Ser262 and Ser356 tau, which are the regulatory sites for tau phosphorylation. These mutations can dramatically reduce the subsequent phosphorylation of several other pathologicallyrelated phosphorylation sites, including PHF-1 (Nishimura et al., 2004).
Using UAS-Tau*S2A and UAS-Aβ* flies, we further analyzed the influence of Aβ on the toxicity of TauS2A. Strikingly, Aβ* co-expression did not worsen the phenotype of Tau*S2A flies. The rough eye phenotype of Tau*S2A flies was not affected when Aβ* was co-expressed. As a comparison, the rough eye phenotype of Tau* flies was more severe (Figure 3A and B).Moreover, when the fly climbing ability was analyzed, the climbing abilities of Aβ*Tau*S2A and Aβ* flies were similar (Figure 3C), suggesting that Aβ was unable to worsen the toxicity of TauS2A. Results from fly longevity assays also supported this conclusion. The lifespan of Aβ* flies was similar to that of Tau*S2A and Aβ*Tau*S2A flies (Figure 3D). The above results indicated that the hypophosphorylated tau mutant was unaffected by Aβ. TauS2A could not execute the toxicity of Aβ, which suggests hyperphosphorylated tau is a critical mediator of Aβ toxicity.
The influence of Aβ on tau toxicity is partly dependent on JNK
Although several kinases and phosphatases control tau phosphorylation, the kinase that mediates the linear interaction between Aβ and tau is still unclear.JNK is one of the kinases activated by Aβ and contributes to tau Ser262 and PHF-1 phosphorylation (Yao et al., 2005; Tatebayashi et al., 2006; Dias-Santagata et al., 2007; Ploia et al., 2011). Therefore, we suspected that JNK may mediate the effects of Aβ on tau protein. This indeed appeared to be the case. First, western blots demonstrated that Aβ* co-expression activated JNK in Tau* flies (Figure 4A and B). Aβ40peptide treatment also activated JNK in Tau*-RFP cells (Additional Figure 2A and B). Second, when SP600125 was used to inhibit JNK activity, we found that JNK inhibition significantly alleviated the severe rough eye phenotype of Aβ*Tau* flies (Figure 4C and D). Similar treatment blocked JNK activation in Aβ* flies (Figure 4E and F) and Aβ-induced tau hyperphosphorylation (Figure 4G and H), indicating that the inhibition of JNK blocked Aβ-induced tau toxicity. Our results suggest that JNK is one of the kinases that mediates the linear interaction between Aβ and tau.
Tau increases Aβ accumulation in the fly model
To further investigate the interaction between Aβ and tau toxicity, we tested the levels of soluble and insoluble Aβ in Aβ*Tau* flies. Surprisingly,compared with levels in Aβ* flies, levels of soluble and insoluble Aβ were elevated in Aβ*Tau* flies (Figure 5A and B), indicating that tau aggravated Aβ accumulation. We further tested whether Aβ degradation was interrupted.The proteases neprilysin 1-3 (NEP1, 2, 3), endothelin-converting enzyme 12(ECE12), angiotensin-converting enzyme (ACE), matrix metalloproteinases(Mmps), and insulin-degrading enzyme (IDE) have been reported to be involved in the Aβ degradation process (Qiu et al., 1998; Carson and Turner,2002; Miners et al., 2008). Therefore, we evaluated the mRNA expression levels of Aβ-degrading enzymes in the flies (Additional Table 1). The RT-PCR results showed that the expression levels ofDrosophilaNep1,Nep3,Mmp2,Nepl7, andIDEwere reduced in Aβ*Tau* flies (Figure 5C and D), while the levels ofNep2,Mmp1,Nepl21, andAncewere unchanged (Figure 5C and D).These results indicated that tau intensified Aβ accumulation, which correlated with the inhibition of the expression of Aβ-degrading enzymes.

Figure 1 |Amyloid-beta (Aβ) expression enhances tau protein toxicity in the Drosophila model.(A) Images of the compound eyes from Tau* flies [GMR-Gal4>UAS-Tau(R406W)].GMR-Gal4 was used to drive Tau* and Aβ* expression in the eyes. Scale bar: 100 μm. (B) Quantitation of rough eye phenotypes in panel A. +++: severe rough eye; ++:medium rough eye; +: mild rough eye; -: normal eye. The number of flies used for eye phenotype calculations was as follows: wild-type (WT) flies, 50; Aβ(Arc2E) flies (Aβ*),38; Tau(R406W) flies (Tau*), 36; Aβ(Arc2E) and Tau(R406W) co-expression (Aβ*Tau*),44 flies. (C) Scanning electron microscopy (EM) images of the compound eyes from Tau*and Aβ*Tau* flies. Scale bar: 100 μm. For each genotype, four adult flies were randomly chosen and quickly frozen in liquid N2. Scanning-EM images were collected under the following conditions: voltage, 15 kV; pressure, 300 Pa; 4°C with 36.9% humidity. For Figure 1C, scanning-EM images of the rough eye phenotype are shown. The rough eye phenotype was observed and evaluated according to the fusion of ommatidia, dents in the retina, and reduced eye size. (D) The lifespans for Tau* flies and Aβ*Tau* flies. ELAVGal4 was used to drive Tau* and Aβ* expression in the fly central nervous system (CNS).The number of flies used for lifespan analyses: Aβ*Tau* flies, 102; Aβ* flies, 114; Tau*flies, 108; WT flies, 116. (E) Climbing ability of different lines of flies. ELAV-Gal4 was used to drive Tau* and Aβ* expression in the CNS. Data are presented as the mean ± SEM. *P< 0.05, **P < 0.01, ***P < 0.001 (unpaired t-test).
Our results indicated that the toxicity of Aβ and tau was reciprocally aggravated. The toxicity of Aβ was dependent on tau hyperphosphorylation.Aβ expression resulted in the hyperphosphorylation and accumulation of tau via JNK activation. Tau also blocked the mRNA expression of Aβdegrading enzymes, such asdNep1,dNep3,dMmp2,dNep4, anddIDE, which subsequently exacerbated Aβ accumulation and toxicity (Figure 5E). This interaction loop will further deteriorate AD pathology.
Discussion
At the early stage of AD, Aβ is considered to be an initiating element (Bloom,2014), which triggers the dysfunction of synapses. Subsequently, normal tau proteins are converted into hyperphosphorylated and pathological forms,leading to changes in tau localization, distribution, and aggregation (Zempel et al., 2010). In this cascade, tau proteins act as the downstream effectors of Aβ toxicity. Increasing evidence indicates that the toxicity of Aβ is largely dependent on tau (Bloom, 2014). Tau pathology correlates well with the neurodegenerative process and cognitive decline in AD, and, typically, tau deficiency can substantially eliminate Aβ toxicity in animal models (Ittner et al., 2010; Ossenkoppele et al., 2016). Our study further suggests that Aβ toxicity is dependent on tau hyperphosphorylation. Therefore, AD may be considered as a form of Aβ-facilitated tauopathy: Aβ triggers tau dysregulation and toxicity (van der Kant et al., 2020).
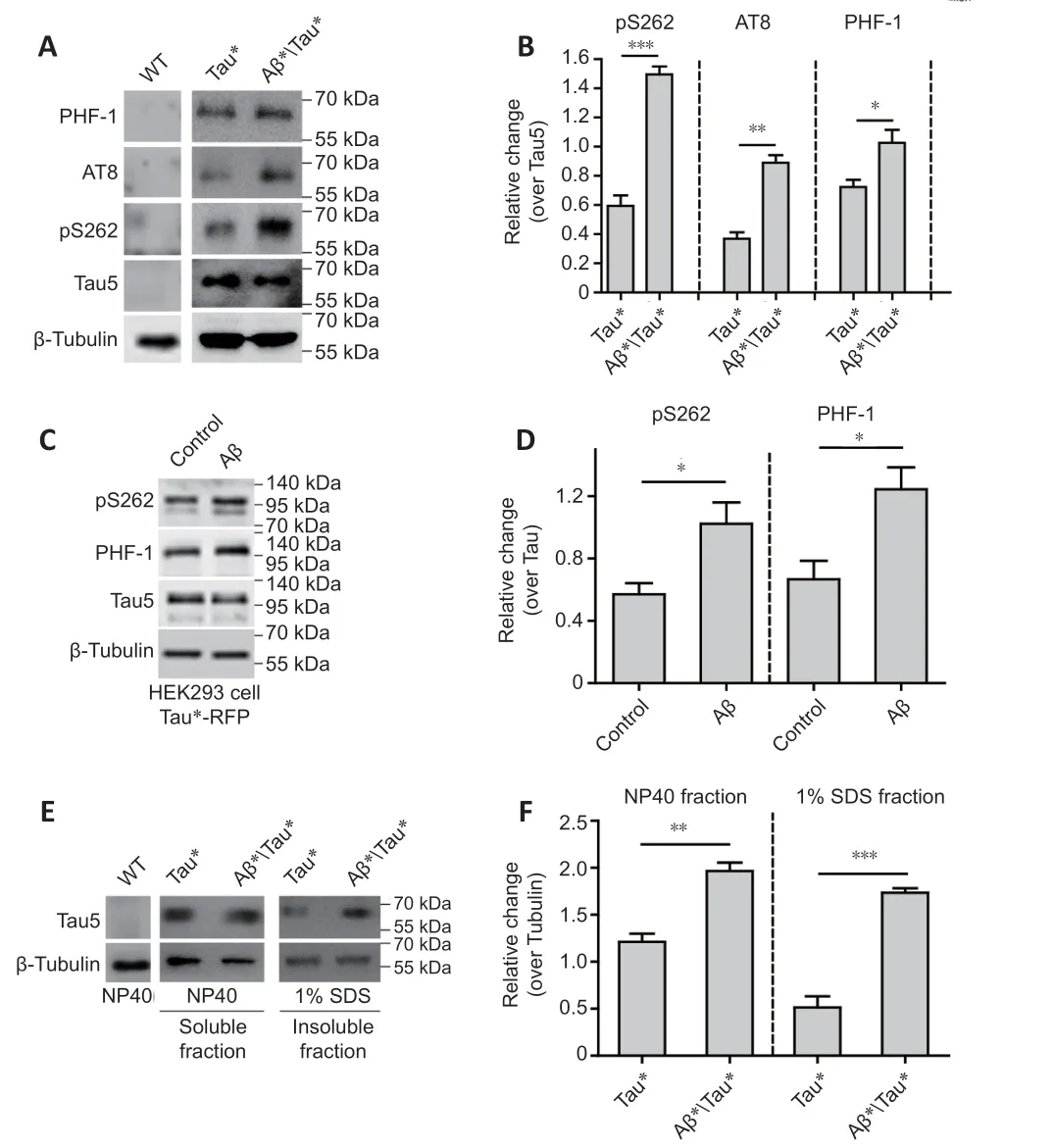
Figure 2 |Amyloid-beta (Aβ) aggravates tau protein hyperphosphorylation and accumulation.(A) Western blot analysis of tau phosphorylation using phosphorylation site-specific antibodies. Protein extracts were prepared from 25 adult fly heads. ELAV-Gal4 was used to drive Tau* and Aβ* expression in the fly central nervous system (CNS). Tubulin was used as a loading control. (B) The quantitative results for panel A. The pS262, PHF-1, and AT8 levels were normalized to the Tau-5 antibody signal. (C) Western blot analysis of tau hyperphosphorylation in HEK293 cells with Tau*-RFP expression. Tau*-RFP cells were treated with 10 μM Aβ40 peptide for 24 hours. Tubulin was used as the loading control. (D)The quantitative results for panel C. Western blot results were calculated using ImageJ software. Levels of pS262 and PHF-1 were normalized to tau5. (E) Western blot analysis of the levels of soluble and insoluble tau protein. Tubulin was used as the loading control.Protein extracts were prepared from 50 adult fly heads. ELAV-Gal4 was used to drive Tau* and Aβ* expression in the fly CNS. (F) The quantitative results for panel E. The tau protein level was normalized to tubulin. Data are presented as the mean ± SEM. *P < 0.05,**P < 0.01, ***P < 0.001 (unpaired t-test). All western blot analyses were performed three times, and representative blots are shown.
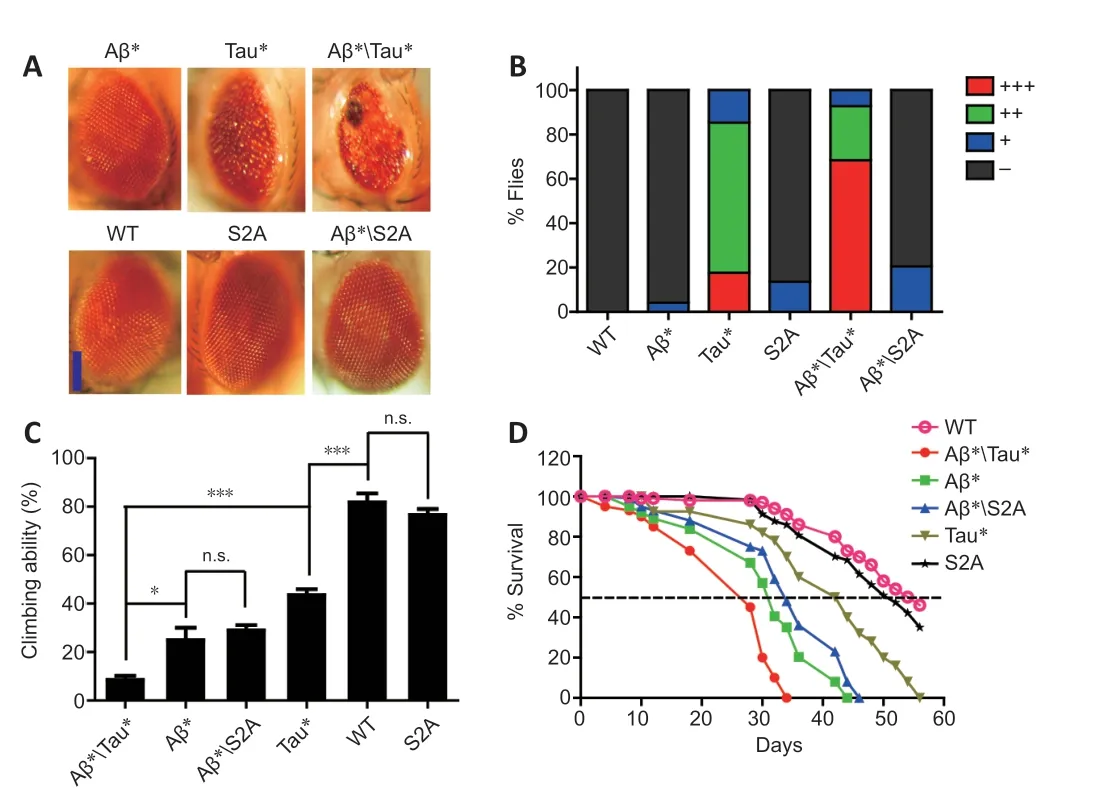
Figure 3 |Hypophosphorylated mutant tau protein is resistant to amyloid-beta (Aβ).(A) Images of fly compound eyes. GMR-Gal4 was used to drive Tau(R406W) (designated as Tau*), Tau*(S2A), and Aβ(Arc2E) (designated as Aβ*) expression in fly eyes. Scale bar: 100 μm. (B) The quantitative results for rough eye phenotypes presented in panel A. +++: Severe rough; ++: medium rough; +: mild rough; -: normal. The number of flies used for eye phenotype calculations: wild-type (WT) flies, 50; Aβ* flies, 48; Tau* flies,34; Tau*(S2A) flies, 44; Aβ*Tau* flies, 41; Aβ*Tau*(S2A) flies, 44. (C) Climbing ability of Tau*(S2A) and Tau* flies. ELAV-Gal4 was used to drive Tau*(S2A), Tau*, and Aβ*expression in the fly central nervous system (CNS). Data are presented as the mean ±SEM. *P < 0.05, **P < 0.01, ***P < 0.001 (unpaired t-test). (D) Lifespans of Tau* and Tau*(S2A) flies. ELAV-Gal4 was used to drive Tau*, Tau*(S2A), and Aβ* expression in the fly CNS.
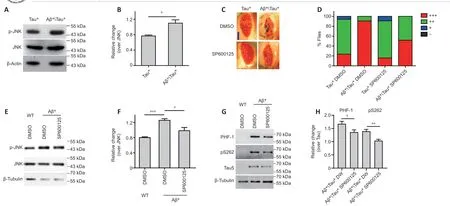
Figure 4 |Amyloid-beta (Aβ) aggravates tau protein toxicity via JNK activation.(A) Representative western blot (WB) showing the level of phosphorylated (p) c-Jun N-terminal kinase (JNK). Protein extracts were prepared from 25 adult fly heads; ELAV-Gal4 was used to drive Tau(R406W) (designated as Tau*) and Aβ(Arc2E) (designated as Aβ*) expression in the fly central nervous system (CNS). Actin was used as the loading control. (B)The quantitative results for panel A; p-JNK level was normalized to JNK. (C) Images of fly compound eyes. GMR-Gal4 was used to drive Tau* and Aβ* expression in fly eyes; 10 μM SP600125 was used to inhibit JNK activity. Scale bar: 100 μm. (D) The quantitative results for panel C. +++: severe rough eye; ++: medium rough eye; +: mild rough eye; -: normal eye.The number of flies used for eye phenotype calculations: DMSO-treated Tau* flies, 34; SP60015-treated Tau* flies, 32; DMSO-treated Aβ*Tau* flies, 44; SP600125-treated Aβ*Tau*flies, 29. (E) Representative WB analysis of the level of p-JNK in SP600125-treated Aβ* flies. Protein extracts were prepared from 25 adult fly heads. ELAV-Gal4 was used to drive Aβ*expression in the fly CNS. Tubulin was used as the loading control. (F) The quantitative results for panel E; the p-JNK level was normalized to JNK. (G) Representative WB analysis of tau hyperphosphorylation in SP600125-treated flies. Protein extracts were prepared from 25 adult fly heads. ELAV-Gal4 was used to drive Tau* and Aβ* expression in the fly CNS. Tubulin was used as the loading control. (H) The quantitative results for panel G; pS262 and PHF-1 signals were normalized to the Tau-5 antibody signal. All WBs were repeated three times.Data are presented as the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 (unpaired t-test).
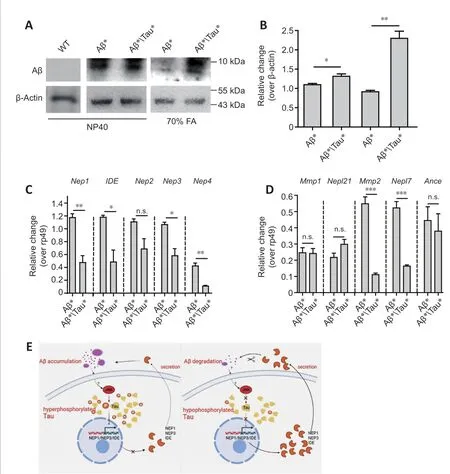
Figure 5 |Tau reduces amyloid-beta (Aβ) degradation in the Drosophila model.(A) Soluble and insoluble Aβ levels were analyzed by western blot using an anti-Aβ antibody. Actin was used as the loading control. ELAV-Gal4 was used to drive Tau* and Aβ* expression in the fly central nervous system (CNS). Western blots were repeated three times, and a representative blot is shown. (B) Quantitative results for panel A;the Aβ level was normalized to actin. (C and D) RT-PCR analysis of the mRNA expression levels of genes encoding Aβ-degrading enzymes (Additional Table 1); dNep1, dNep2,dNep3, dNep4, dIDE, dMmp1, dMmp2, dNepl7, dNepl21 and dAnce mRNA levels were analyzed by RT-PCR, and rp49 was used as the loading control. Statistical differences were determined from the band intensities, which were analyzed by ImageJ software.The intensity of each band was normalized to the intensity of rp49. ELAV-Gal4 was used to drive Tau* and Aβ* expression in the fly CNS. Data are presented as the mean ± SEM.*P < 0.05, **P < 0.01, ***P < 0.001 (unpaired t-test). n.s.: Not significant. (E) A model depicting the reciprocal interaction between Aβ and Tau toxicity. 1) Toxicity of Aβ is dependent on tau hyperphosphorylation. 2) The hypophosphorylated tau mutant is not affected by Aβ. Aβ accumulation activates JNK and results in the hyperphosphorylation and accumulation of tau. Tau can also block the mRNA expression of the Aβ-degrading enzymes Nep1, Nep3, Nep4, Mmp2, and IDE, which may block the clearance of Aβ and result in the aggravation of Aβ accumulation and toxicity. This reciprocal interaction loop further accelerates AD progression and pathology.
To understand the Aβ-tau cascade, it is critical to characterize the mediators.Our results indicated that the Aβ-mediated increase in tau toxicity was partly dependent on JNK. This phenomenon was also found in other models (Wei et al., 2002; Yao et al., 2005; Ma et al., 2009). Importantly,JNK can phosphorylate tau protein on the phospho-epitopes related to AD pathology, including Ser262, Ser396, and Ser404 (Goedert et al., 1997;Yoshida et al., 2004; Cao et al., 2013; Tyagi et al., 2013). JNK is correlated with AD pathology; it is redistributed and activated in AD and is associated with neuronal neurofibrillary tangles (Hensley et al., 1999; Zhu et al., 2001;Yoshida et al., 2004; Lagalwar et al., 2006). In our study, inhibition of JNK activity by SP600125 significantly reduced tau toxicity, which was aggravated by Aβ. Although the Aβ-JNK-Tau cascade can be established, the underlying mechanism of Aβ-induced JNK activation is still unclear. Indeed, JNK can be activated by several stimuli (Mehan et al., 2011). Some of these stimuli are related to AD pathology. For example, oxidative stress can specifically activate JNK and is also prevalent in the AD brain. In our experiments, we used vitamin C to reduce oxidative stress in Aβ co-expressed flies. Vitamin C partially eliminated tau hyperphosphorylation induced by Aβ, which was accompanied by a reduction in JNK activity (Additional Figure 3A and B). These data suggest that oxidative stress was one of the mediators that contributed to Aβinduced JNK activation and tau hyperphosphorylation; however, the detailed mechanism still requires further investigation. Aβ can cause oxidative stress in several other models (Shelat et al., 2008; Cheignon et al., 2018). Oxidative stress may induce the oxidation of Trx1 and JNK inhibitory protein, which in turn may lead to the activation of JNK (Han et al., 2009). Aβ can induce ER stress signaling (Fonseca et al., 2013), which can lead to JNK activation(Fonseca et al., 2013) through activation of the transmembrane protein kinase IRE1 (Urano et al., 2000). DNA damage response pathways can also be activated by Aβ (Santiard-Baron et al., 2001), resulting in subsequent JNK activation (Hamdi et al., 2005). Furthermore, alterations in inflammatory cytokines released from microglia in the AD brain have been reported to activate JNK (Wang et al., 2015).
Our results suggest that Aβ and tau toxicities can be reciprocally aggravated.Aβ can aggravate tau hyperphosphorylation and toxicity, and, reciprocally,tau can aggravate Aβ deposition. This Aβ-JNK-Tau-Aβ loop will accelerate the progression of AD. In fact, supporting our findings in the fly model, the formation of Aβ plaques in mice can also be enhanced by tau (Jackson et al.,2016), and Aβ can significantly promote tau aggregation (He et al., 2018).
Aβ(Arc2E) expression in the fly can cause a severe phenotype as previously described (Crowther et al., 2005), including rough eye and degeneration.However, our results showed that Aβ(Arc2E) expression did not induce a rough eye phenotype, and we suspected that the expression level of Aβ in our Aβ(Arc2E) flies was lower than the previously used fly line. To test this possibility, we expressed Aβ(Arc2E) in fly eyes using high-temperature conditions (29°C). Our results indicated that Aβ(Arc2E) caused a rough eye phenotype at 29°C but not at 25°C; moreover, Aβ(Arc2E) also aggravated the rough eye phenotype at 29°C (Additional Figure 4).
As described, Aβ and tau can work synergistically (Folwell et al., 2010). Aβ expression can synergistically enhance several phenotypes caused by tau expression, such as disruption of axonal transport and the neuromuscular junction related to synaptic dysfunction (Folwell et al., 2010). Aβ and tau protein can interact synergistically through the direct interaction between the R2 domain of tau and Aβ oligomers, leading to the acceleration of protein aggregation (Miller et al., 2011). The synergistic interaction between Aβ and tau was indeed involved in and contributed to the neuropathology in an AD animal model (Clinton et al., 2010).How tau aggravates Aβ accumulation is unclear. Our results indicate that tau can reduce the mRNA expression of genes encoding Aβ-degrading enzymes, includingdNep1,dNep3,dMmp2,dNep4, anddIDE. It is likely that tau can interfere with the processing of Aβ. According to our results,the level ofdBace1mRNA, which encodes an APP cleaving enzyme, was increased, but the mRNA level of the presenilin gene dPsn was reduced when Tau* was co-expressed with Aβ* (Additional Figure 5). However, this result needs to be investigated further. As described, tau protein can be found in the nucleus and is related to heterochromatin stability. The level of nuclear hyperphosphorylated tau is associated with stress stimulation.In particular, the ribosomal DNA transcriptional repression-related protein TIP5 is associated with tau protein, and this association may result in the regulation and suppression of gene transcription (Maina et al., 2018). Tau aggregates can also be associated with U2 snRNA, which can regulate gene expression and harbor transcription-related RNAs and proteins (Galganski et al., 2017). Therefore, besides the disruption of neuronal function, such as the interference in axon transport and neurotransmitter release, pathological tau may also decrease the expression of genes related to Aβ clearance. As a consequence, Aβ and tau can reciprocally intensify the protein accumulation of each other in AD. This reciprocal loop between Aβ and tau further accelerates and aggravates AD and may cause the variation in AD pathology.This reciprocal relationship between Aβ and tau should be considered and investigated for development of future AD treatments.
Limitations
Although some of the results were repeated using the HEK293 cells, the majority of results in the current study were generated from theDrosophilamodel. The mammalian system is more complex thanDrosophila, and our results will require confirmation in higher-order model organisms. We propose that Aβ and tau can be considered as a combined target for drug development. However, the mechanisms by which Aβ induces JNK activation and tau inhibits the expression of genes for Aβ-degrading enzymes will require the use of biochemical and -omics approaches in the future.
Conclusion
By using theDrosophilaAD model, we found that Aβ and tau can reciprocally aggravate their toxicities and promote disease progression through the respective activation of JNK and inhibition of the expression of Aβ-degrading enzymes.
Acknowledgments:We are grateful to the Bloomington Drosophila Stock Center for fly stocks, the Biomedical Analysis Center of Tsinghua University and the Biomedical Analysis Center of Hangzhou Institute for Advanced Study for their help and services, Dr. Bing-Wei Lu of Stanford University and Dr. Yi Zhong of Tsinghua University for their kind gifts of the flies and reagents. We also thank Zhi-Qing Wang and Wen-Jia Qiang of Tsinghua University for their technical helps.
Author contributions:YPH, BZ, and JRW designed the experiments. YPH,ZDS and JXH performed the experiments. YPH analyzed data and wrote the manuscript. All authors approved the final version of the manuscript.
Conflicts of interest:The authors declare that there is no conflict of interest.Availability of data and materials:All data generated or analyzed during this study are included in this published article and its supplementary information files.
Who are you? said Browny, starting up in great fright, for though the voice sounded gentle, he felt sure it was a feigned13 voice, and he feared it was the fox.
Open access statement:This is an open access journal, andarticles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Mohammad Moshahid Khan, University of Tennessee Health Science Center, USA.
Additional files:
Additional file 1: Open peer review report 1.
Additional Figure 1: Approach to co-expression Tau*and Aβ*in fly eyes and CNS.
Additional Figure 2: Amyloid-beta (Aβ)-induced JNK activation in HEK293 cells.
Additional Figure 3: Vitamin C eliminates amyloid-beta (Aβ)-induced tau hyperphosphorylation.
Additional Figure 4: Aβ(Arc2E) causes rough eye phenotype and aggravate tau toxicity at 29°C.
Additional Figure 5: Tau alerts dPsn and dBace mRNA expression in flies.
Additional Table 1: Homologues of amyloid-beta (Aβ)-degrading enzymes on the fly genome.

- 中国神经再生研究(英文版)的其它文章
- Complement activation kindles the transition of acute posttraumatic brain injury to a chronic inflammatory disease
- Role of adipose tissue grafting and adipose-derived stem cells in peripheral nerve surgery
- Potential significance of CX3CR1 dynamics in stress resilience against neuronal disorders
- A comparative analysis of differentially expressed genes in rostral and caudal regions after spinal cord injury in rats
- The delivery of miR-21a-5p by extracellular vesicles induces microglial polarization via the STAT3 pathway following hypoxia-ischemia in neonatal mice
- Basic mechanisms of peripheral nerve injury and treatment via electrical stimulation