
Potential significance of CX3CR1 dynamics in stress resilience against neuronal disorders
2022-03-09 07:34:16KoichiInoue
中国神经再生研究(英文版) 2022年10期
Koichi Inoue
Abstract Recent findings have implicated inflammatory responses in the central nervous system in a variety of neuropsychiatric and neurodegenerative diseases, and the understanding and control of immunological responses could be a major factor of future therapeutic strategies for neurological disorders. Microglia, derived from myelogenous cells, respond to a number of stimuli and make immune responses, resulting in a prominent role as cells that act on inflammation in the central nervous system. Fractalkine (FKN or CX3CL1) signaling is an important factor that influences the inflammatory response of microglia. The receptor for FKN, CX3CR1, is usually expressed in microglia in the brain, and therefore the inflammatory response of microglia is modified by FKN. Reportedly, FKN often suppresses inflammatory responses in microglia and activation of its receptor may be effective in the treatment of inflammatory neurological disorders. However, it has also been suggested that inflammatory responses facilitated by FKN signaling aggravate neurological disorders. Thus, further studies are still required to resolve the conflicting interpretation of the protective or deleterious contribution of microglial FKN signaling. Yet notably, regulation of FKN signaling has recently been shown to be beneficial in the treatment of human diseases, although not neurological diseases. In addition, a CX3CR1 inhibitor has been developed and successfully tested in animal models, and it is expected to be in human clinical trials in the future. In this review, I describe the potential therapeutic consideration of microglial CX3CR1 dynamics through altered FKN signaling.
Key Words: Alzheimer’s disease; CX3CR1; fractalkine; inflammation; knockout mice; microglia;resilience; SARS-CoV-2; stress; stroke
Introduction
“Resilience” refers to the ability to overcome and recover from stressful and/or adverse circumstances (Rutter, 1985; Grotberg, 2003). In addition to personal qualities, resilience can be affected by family, social, and other environmental factors, and it can lead to later onset of disorders caused by factors other than personal qualities, such as neuropsychiatric disorders caused by environmental changes. For example, it has been suggested that economic problems may contribute to 20% of depressions (Dudek et al.,2021), which should be other than personal qualities for many children.On the contrary, some of those disorders can be treated by acquiring stress resilience, which was not previously present (Feder et al., 2019).This suggests that a well-functioning society for the treatment of diseaserelated stress resilience could be the foundation for a healthy life that does not rely on medical costs and care, which is a global problem. Even if not at the level of diseases, this competence is required more than ever in a modern and complicated society where the spread of SARS-CoV-2 infection worldwide has brought economic depression and made it difficult to imagine a promising near future with high expectations. In fact, COVID-19 can cause serious health, social, and economic crises for families, including children(Barzilay et al., 2020; Prime et al., 2020). Stress resilience is not only related to functional mental activity but also to substantial neurological impairment.For example, higher susceptibility to stress has been reported to result in a higher prevalence of stroke and to affect not only stroke onset but also poststroke complications and recovery (Surtees et al., 2007; Bergh et al., 2014).Resilience also plays an important role in the pathogenesis of not only stroke but also dementia and other hereditary disorders (Bergman et al., 2010; Maul et al., 2020). In line with this, animal studies have clearly reported that stress during the perinatal period through to childhood influences the function of the hypothalamic-pituitary-adrenal axis, leading to a decrease in stress resilience in later life, and further understanding of stress resilience is an important issue for a better future (Liu et al., 1997; Sapolsky, 1997).
Thus, the term “resilience” or “stress resilience” has become very common in scientific papers over the past decade or so. For instance, a PubMed search for “stress resilience” retrieves 37 papers in 2000, 356 in 2010, and 2190 in 2020. This may also be due to the social and medical trend of considering treatment and recovery from diseases not only from the physical but also mental aspects. In response to the question of how resilience is imparted,psychologists sometimes speak of mental strength such as endurance and perseverance, but its mechanisms have not yet been clarified at the molecular level in the field of life science (Feder et al., 2009; Menard et al., 2017). In this regard, we have recently focused on fractalkine (FKN)/CX3CR1 signaling in microglia and found a phenomenon that may be generally involved in the resilience of function of the central nervous system (Inoue et al., 2021).Considering the findings, in this review, I will give an overview of FKN and its signaling in microglia and review its possible significant involvement in the universal neuropsychiatric activity.
Search Strategy and Selection Criteria
Studies cited in this narrative review published from 1985 to 2021 were searched on the PubMed database using the following keywords: fractalkine,CX3CR1, stress, resilience, microglia, inflammation, stroke, Alzheimer’s disease, SARS-CoV-2, knockout mice.
Fractalkine and Its Receptor CX3CR1
FKN has been identified as a chemokine that exerts chemotactic activity on certain cells. Its receptor, CX3CR1, is expressed mainly in immune cells,such as macrophages and microglia, but it is also present in various other cells, such as neurons, astrocytes, and vascular endothelial cells (Meucci et al., 2000; Umehara et al., 2001; Limatola and Ransohoff, 2014; Lee et al.,2018). CX3CR1 is a G protein-coupled receptor, and its activation leads to Ca2+influx and change in cAMP level, followed by transduction of downstream signals regarding cell differentiation and survival, such as MAPKs and Akt/PKB(Maciejewski-Lenoir et al., 1999; Meucci et al., 2000; Kansra et al., 2001; Davis and Harrison, 2006; Dorgham et al., 2009). Its expression level is demonstrated to be increased by external stimuli in some cells (Maciejewski-Lenoir et al., 1999; Fong et al., 2000; Sung et al., 2005; O’Sullivan et al., 2016; Ho et al., 2020; Kawamura et al., 2020), and the extent of its expression change contributes to changes in the intensity of the signal downstream of CX3CR1, as does the release of the ligand, FKN.
FKN is released from various types of cells, including neuronal, vascular endothelial, intestinal, and epithelial cells. While the stationary release is recognized, the amount of release is altered by stimuli. In both cases,FKN acts as a paracrine factor on the peripheral cells and as a hormone remotely (Dreymueller et al., 2012; Lee et al., 2018). Thus, FKN is a released protein that is initially translated as a membrane surface protein with a transmembrane region (Bazan et al., 1997; Fong et al., 2000; Poniatowski et al., 2017; Lee et al., 2018). It is excised at the region slightly closer to the N-terminal side from the transmembrane domain by serine proteases, such as ADAM17 and ADAM10, and released into the bloodstream. This soluble FKN accounts for FKN blood level. Apart from the soluble form, FKN often remains uncut and unreleased and acts on the cellular membrane. For example,CX3CR1-expressing macrophages or monocytes are trapped on membranebound (uncut) FKN-producing vascular endothelial cells, using them as a scaffold to assist penetration and migration of CX3CR1-positive cells (Bazan et al., 1997; Fong et al., 2000). As a timely topic, it has been reported that angiotensin II causes an increase in FKN expression in vascular endothelial cells (Rius et al., 2013). In addition, it has been suggested that FKN may play a role in severe SARS-CoV-2 infections caused by cytokine storms because of the relationship between angiotensin II and an angiotensin II-degrading enzyme ACE2, which is a target of the SARS-CoV-2 protein (Banu et al., 2020;Gracia-Hernandez et al., 2020; Rivas-Fuentes et al., 2021).
Current Status of FKN Signaling in Human Diseases
Genetic mutations of human CX3CR1 are associated with the risk of cardiovascular and psychiatric diseases, such as schizophrenia. For example,mutation of T280M in FKN makes less activation of Ca2+influx-mediated cell signaling compared to wild-type FKN, resulting in weakened adhesion and invasion of CX3CR1-positive leukocytes into the vascular wall. This reduces cardiovascular diseases, such as atherosclerosis (McDermott et al., 2003). In addition, mutation of A55T in FKN attenuates at least Akt signaling, and the changes in the signaling strength may modulate cellular function, leading to altered brain functions or development of diseases (Ishizuka et al., 2017).
Considering that attenuated activity of FKN signals decreases disease incidence, increased activation can enhance disease development. Other than mutation, recent findings have revealed that FKN signals are abnormally evoked in human diseases such as rheumatoid arthritis, Crohn’s diseases,and systemic scleroderma, and the FKN neutralizing antibody therapy may be functional (Muehlhoefer et al., 2000; Ruth et al., 2001; D’Haese et al., 2012;Luong et al., 2019; Matsuoka et al., 2021; Tanaka et al., 2021a, b). In fact,a phase II clinical trial has been conducted using FKN neutralizing antibody in rheumatoid arthritis patients who did not respond well to methotrexate,a clinically commonly used drug. Although the patients were treated for as short as 6 months, there seemed to be a significant effect on moderation(Tanaka et al., 2021a). These reports explore the possibility that modulation of FKN signaling could become curable therapeutic strategies for human diseases.
Dynamics of Microglial CX3CR1 and Its Effects on Neuronal Function
As mentioned above, FKN signals play critical roles in the modulation of neuronal function. The deficiency of FKN has often resulted in neurological defects, but their phenotypes are not so severe (Cook et al., 2001;Sokolowski et al., 2014; Winter et al., 2020). CX3CR1-deficient mice have also been reported to have a variety of neurological problems. For example,inflammation-induced neurotoxicity in stroke models is reduced in CX3CR1-deficient mice, whereas neurotoxicity appears to be increased by CX3CR1-deficiency in Parkinson’s disease and amyotrophic lateral sclerosis (ALS)models (Cardona et al., 2006; Tang et al., 2014; Wang et al., 2018). A relationship with stress resilience has also been reported in CX3CR1-deficient mice, in which the deficiency reduces stress-induced memory and cognitive impairment and alleviates depressive symptoms (Rimmerman et al., 2017; Liu et al., 2020).
Indeed, phenotypes exhibited in CX3CR1-deficient mice could be referable.However, it is almost impossible for real animals to lose the expression of CX3CR1 to the same extent as knock-out mice do, and therefore it is reasonable to presume that the effect of signaling intensity is attributable to changes in CX3CR1 expression level. Tables 1 and 2 list examples of reports regarding changes in microglial CX3CR1 expression levels (both protein and gene expressions) due to various stresses since it would not be meaningful to describe details of knockout mice. In the tables, brain regions (Table 1) and cell types (Table 2), in which CX3CR1 changes, are described along with stimuli and models that cause these changes and with the subsequent results. Detection methods are also mentioned. From these tables, it should be understood that CX3CR1 is altered by a variety of stimuli, which in turn changes the phenotypes, including neurological symptoms. As shown in Table 1, while the stimuli affecting the expression are not identified in somein vivodata, responsive stimuli are mostly identified, and the available literature implies that a variety of stimuli can modify the expression of CX3CR1. Of note,promoter analysis of humanCX3CR1has been performed, and the importance of the family of transcription factors, nuclear factor of activated T cells, has been suggested (Barlic et al., 2004). Nuclear factor of activated T cells are mainly present in immune, cardiovascular, neuronal, and muscular cells. They are dephosphorylated by calcineurin, a phosphatase that is activated through intracellular Ca2+elevation, resulting in nuclear translocation and activation of downstream target gene expression (Hogan et al., 2003; Fric et al., 2012).However, since it is activated by stimuli, such as lipopolysaccharide (LPS) and enhances gene expression (Nagamoto-Combs and Combs, 2010; Fric et al.,2012), it would not play a leading role in the response to LPS stimulation,which decreases the expression level of CX3CR1, as reported by several groups, including ours (Zujovic et al., 2000; Wynne et al., 2010; Wang et al.,2020; Inoue et al., 2021). In addition, downstream effects, such as the survival of circumferential photoreceptors or neuronal cells, are reported (Roche et al., 2016; Wang et al., 2020). Intriguingly, the effects of CX3CR1 regulation on disease mechanisms are likely to be true if consistent phenotypic trends are displayed between models with down-regulation of CX3CR1 and CX3CR1-deficient animals. For instance, accumulation of β-amyloid is decreased in CX3CR1-deficient mice (Lee et al., 2010), and this corresponds to Keren-Shauel’s findings, as shown in Table 1 (Keren-Shaul et al., 2017), suggesting that activation of FKN signaling may exacerbate symptoms in Alzheimer’s disease. Their report indicates that attenuation of FKN signaling intensity in local microglia around the lesion makes them deviated from checkpoints that inhibit unwanted immune responses, leading to an increase in immunological activities including phagocytosis of β-amyloid. However, on the other hand,Gonzalez-Prieto et al. (2021) have reported that Alzheimer’s patients have a higher expression of CX3CR1, and therefore further investigation would be expected. They have also noted microglia with lower expression of CX3CR1 accumulated in the lesion of ALS model mice. On the contrary, ALS model derived from CX3CR1-deficient mice show a more intense immune response,resulting in higher lethality. There is one thing of concern, and at first glance of the tables, it appears that CX3CR1 is often decreased by stimulation in cultured cells, while it is still increased in variousin vivomodels. Although the reason for this is unclear, it has been reported that the expression level of CX3CR1 itself is much higher in human microglia than in cultured cells(Gosselin et al., 2017), and the analysis of CX3CR1 expression in microglia may not be meaningful without usingin vivoreports. Therefore, it is still controversial whether changes in the CX3CR1 level are beneficial. To resolve this, recently, a CX3CR1 antagonist has become commercially available(Karlström et al., 2013; Cederblad et al., 2016; Ho et al., 2020), and changes in phenotypes can be referrable by the antagonist. The next step is to apply this to human diseases. Most of the data shown in Table 1 are derived from animal models, and there is no guarantee that the dynamics of CX3CR1 are consistent between humans and those models. Nevertheless, if they are similar, human phenotypes such as multiple sclerosis and neurogenic hypertension could be treatable with the modification of CX3CR1 function(Ridderstad Wollberg et al., 2014; Ho et al., 2020). To do this hereafter,changes in CX3CR1 should be examined in human microglia. If CX3CR1 in macrophages in the peripheral blood and microglia fluctuates in the same way as SGK1 does in response to glucocorticoids (Anacker et al., 2013), then the change in CX3CR1 in the brain microglia by any stimuli can be reflected in peripheral blood, which may be used to understand and treat CX3CR1-oriented human diseases with lower invasiveness.
Conclusion
The term “stress resilience” has become more common in medical biology papers in the last decade or so. It appears to be related not only to the development of neuropsychiatric disorders but also to exacerbations of Alzheimer’s disease and post-stroke neurodegeneration, and its detailed molecular pathology is expected to be elucidated for the treatment of neurological diseases. FKN signaling is a signal involved in the immune response of microglia that affects stress resilience. Microglia express an FKN receptor, CX3CR1, and exchange signals with ligand-expressing neurons and other cells. The amount and form of the ligand FKN (membranebound or soluble) can inflect the stimulation of microglia and modulate its effects, ultimately modifying brain and nerve functions. Changes in CX3CR1 expression in microglia have been reported often, and it is generally accepted that its expression is altered by various stimuli, which may also occur in human diseases. At the level where mental function is involved, there have been no studies utilizing neutralizing antibodies or the inhibitor, but as in the case of rheumatoid arthritis mentioned above, its increased activity may lead to exacerbation of the condition. We found that the expression level of CX3CR1 in microglia is reduced by stimulation with alcohol or LPS, albeit in cultured cells (Inoue et al., 2021). Since it is often reported that stimulation of CX3CR1 suppresses microglial inflammatory responses in common LPSbased microglial immune research, we consider that if a similar situation occursin vivoand in humans, various stresses may cause suppression of CX3CR1 expression and the consequent FKN signaling function in microglia,thereby reducing the ability of microglia to suppress runaway. Unfortunately,however, there is currently no clear direction as to whether this variation is uniformly good or bad for certain conditions. Nonetheless, this also being the case in humans, if we can prove beneficial changes in CX3CR1 for each individual disease by adjusting the life environment outside of clinical treatment, such as diet and exercise, then we may be able to acquire beneficial stress resilience through FKN signaling.
Author contributions:KI designed, wrote this review, and approved the final version of the manuscript.
Conflicts of interest:There are no conflicts of interest.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
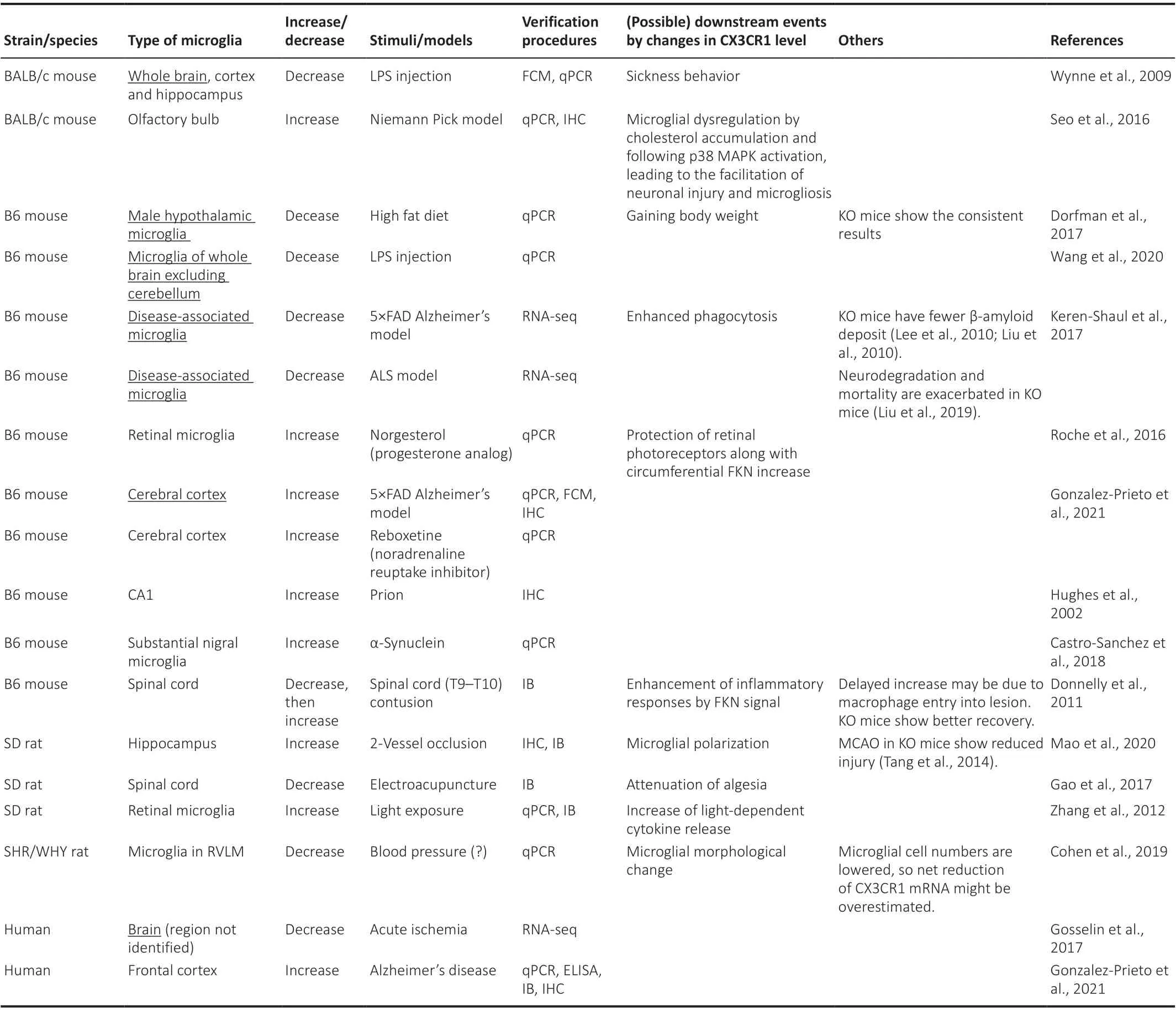
Table 1 |Dynamics of microglial CX3CR1 by stimuli in vivo
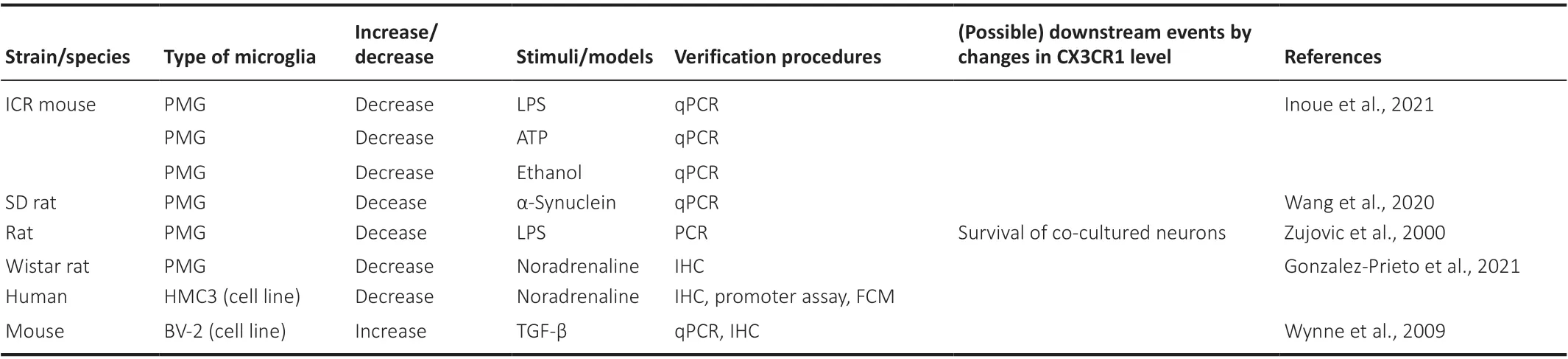
Table 2 |Dynamics of microglial CX3CR1 by stimuli in vitro

- 中国神经再生研究(英文版)的其它文章
- Complement activation kindles the transition of acute posttraumatic brain injury to a chronic inflammatory disease
- Role of adipose tissue grafting and adipose-derived stem cells in peripheral nerve surgery
- Toxicities of amyloid-beta and tau protein are reciprocally enhanced in the Drosophila model
- A comparative analysis of differentially expressed genes in rostral and caudal regions after spinal cord injury in rats
- The delivery of miR-21a-5p by extracellular vesicles induces microglial polarization via the STAT3 pathway following hypoxia-ischemia in neonatal mice
- Basic mechanisms of peripheral nerve injury and treatment via electrical stimulation