
辐射改性介孔SiO2及其对铼的吸附性能
2022-03-05 11:11沈旺来陈怡志吴志豪翁汉钦林铭章
辐射研究与辐射工艺学报 2022年1期
沈旺来 陈怡志 张 鹏 吴志豪 翁汉钦 林铭章
(中国科学技术大学核科学技术学院 合肥 230026)
作为低碳排放的清洁能源,核电正处于高速发展之中,截止2018 年,核电占全球总发电量的10.2%[1-2]。大规模建设的核电站面临自然灾害引发的核事故风险,以及潜在大量放射性废物的释放风险。例如,2011 年福岛核事故向环境中释放(6.3~7.7)×1017Bq 放射性核素,其中,长半衰期的锝-99(99Tc)(2.13×105a)是一种主要的放射性核素[3-4]。作为Tc 的主要存在形式,高锝酸根(TcO4-)与硫酸根具有相似结构,具有极高的水溶性,从而极易进入生物圈,对人体造成巨大的危害[5]。因此,实现对放射性污水中Tc 的高效去除具有重要的意义。
Tc 的所有同位素均具有放射性,这极大限制了Tc的分离研究,因此与Tc具有相似化学性质的非放射性元素铼(Re)常被作为Tc 化学类似物用于研究Tc的分离过程[6-7]。目前,分离Re(Tc)的常用方法包括溶液萃取[8]、吸附[9]等。吸附法在处理大量含有低浓度金属离子的污水时具有巨大的优势,因此被广泛应用于放射性污水的处理[10]。福岛核事故后,日本使用Kurion、SARRY、ALPS等系统去除放射性污水中的62 种放射性核素,其中十六烷基三甲基铵改性的沸石被用于吸附废水中的Tc[11]。然而改性后的沸石表面所带的负电荷与TcO4-之间的静电排斥导致对其对Tc的吸附容量仍然较低[12-13]。而离子交换树脂等有机吸附材料对Tc 虽然有较高的吸附容量,但其仍然存在吸附速率低的缺点[14]。目前对放射性污水中Tc的快速高效的去除仍是一个亟需解决的问题。
吸附剂的官能团是影响吸附过程的决定性因素,近年来冠醚[15]、哌嗪[16]和含氮杂环[17]等官能团被用于Tc 的吸附,但其高额的成本和复杂的合成过程极大地限制了这些基团的应用。而在吸附放射性核素的过程中,吸附剂中官能团的辐射稳定性也是限制吸附剂吸附性能的重要因素。吡啶基团由于其较低的成本和强辐射稳定性,在去除Tc 方面具有独特的优势[18-19]。Zu 等合成的聚4-乙烯基吡啶改性的聚苯乙烯微球对Re(Tc)具有良好的吸附性能,但其中的叔胺只有在酸性下才能质子化达到吸附ReO4-和TcO4-的目的,这极大限制了其对环境中放射性污水的处理[20]。当吡啶基团中的N 被季铵化改性后,基团固有的正电荷使吸附剂在近中性下仍能对ReO4-具有高效的吸附性能[21]。而利用含有多个正电位点的基团对吡啶季铵化改性则可以进一步提高吸附剂对Re(Tc)的吸附容量。
除官能团外,吸附剂的孔结构对吸附过程也具有重要的影响。大孔(或较大的介孔)结构可以有效促进传质过程,而具有大比表面积的微孔(或较小的介孔)结构则可以提供大量的吸附位点。结合了大孔(或较大的介孔)和微孔(或较小的介孔)的多层次孔结构兼具了快的传质速率和比表面积[22],Yang 等[23]发现同时具有约12.5 nm 和2.8 nm 介孔的多层次介孔SiO2(F-SiO2)经改性后对放射性核素具有较快的吸附速率和高的吸附容量。基于此,季铵化吡啶基团改性后的F-SiO2在Re(Tc)的高效吸附方面具有巨大的应用前景。
在本文工作中,我们以合成F-SiO2为基材,对其进行4-VP的辐射接枝,并优化了多种辐射参数,以达到最大的接枝率,然后对优化后的样品进行季铵化,进一步考察其对Re(VII)的各项吸附性能。研究结果表明,所合成的季铵化F-SiO2(FSVPQ)对去除放射性污水中的Tc 具有较高的应用价值。
1 材料与方法
1.1 原料与试剂
甲基丙烯酸-3-(三甲氧基甲硅烷基)丙酯(C10H20O5Si,MPS,98%,含稳定剂BHT)、2-溴乙基三甲基溴化铵(C5H13Br2N,2-BETAB,99%)和4-乙烯基吡啶(C7H7N,4-VP,95%,含稳定剂TBC)购于梯希爱(上海)化成工业发展有限公司;十六烷基三甲基溴化铵(C19H42BrN,CTAB,99%)购于百灵威科技有限公司;二甲基甲酰胺(C3H7NO,DMF,99.5%)购于成都市科隆化学品有限公司;尿素(CH4N2O,Urea,99%)购于西格玛奥德里奇(中国)有限公司;正硅酸四乙酯(C8H20O4Si,TEOS,99%)购于萨恩化学技术(上海)有限公司;高铼酸钠(NaReO4,99%)购于阿法埃莎(中国)化学有限公司;0.4 nm 分子筛、硝酸钠(NaNO3)、硫酸钠(Na2SO4)、氯化钠(NaCl)、七水合硫酸亚铁(FeSO4·7H2O)、盐酸(HCl)、二水合草酸(H2C2O4·2H2O)、甲醇(CH3OH)和二甲苯(C8H10)、异丙醇(C3H8O,IPA)、环己烷(C6H12)均为分析纯试剂,购于国药集团化学试剂有限公司。含1.0 mol/L HNO3的铼单元素标准溶液(1 000 μg/mL)购于国家有色金属及电子材料分析测试中心。实验中使用的去离子水(电阻率为18.25 MΩ·cm)由卡尔顿(中国)水务有限公司的Kertone Lab Vip 超纯水机处理。DMF 先与氢化钙混合加热回流去除水分,再蒸馏收集,保存时加入经550 ℃活化的0.4 nm分子筛。
1.2 辐射法制备吡啶功能化F-SiO2
1.2.1双键改性F-SiO2的制备
取5.0 g CTAB 和3.0 g 尿素加入圆底烧瓶内,再加入150 mL水搅拌至溶解。然后加入150 mL环己烷与4.6 mL异丙醇,并逐滴加入12.5 mL TEOS,在40 ℃下保持搅拌30 min。升温至70 ℃后反应16 h,同时使用直型球形冷凝管进行回流。反应结束后加入150 mL 无水乙醇破坏乳液结构,将混合液置于离心管中离心收集固相产物,离心参数为10 000 r/min,持续5 min。用乙醇和超纯水分别洗涤固体产物3 次后,在60 ℃的真空烘箱中干燥24 h。最终将粗产品置于坩埚中,于马弗炉550 ℃下煅烧5 h,以去除CTAB 和其他有机杂质,样品命名为F-SiO2。
取0.3 g F-SiO2加入圆底烧瓶中,再加入22.5 mL二甲苯和2.5 mL MPS,将固体超声分散后,通氮气15 min,于110 ℃下反应8 h。固体经离心收集(1 000 r/min,5 min),再用乙醇洗涤5 次后于60 ℃真空烘箱中干燥6 h。样品命名为FS-MPS。
1.2.2聚4-乙烯基吡啶功能化F-SiO2的制备
取0.3 g FS-MPS在冰水浴下超声分散至已纯化的12.5 mL 4-VP 中,取50 mg FeSO4·7H2O 溶 于12.5 mL甲醇中。将上述甲醇溶液与4-VP在冰水浴下混合并超声,通氮气15 min 后置于60Co(7.4 ×1014Bq)辐射场中,照射一段时间后取出。产物经离心收集(1 000 r/min,5 min)后使用10%草酸溶液、甲醇和乙醇分别洗涤3 次,于60 ℃真空烘箱中干燥6 h。样品命名为FS-VP。
1.2.3季铵化FS-VP的制备
取0.3 g FS-VP 溶于25 mL DMF中,加入0.5 g 2-BETAB 并用铝箔纸包实圆底烧瓶以防止卤化物见光分解[24],升温至60 ℃反应3 d。产物经离心收集(1 000 r/min,5 min)后使用DMF 和乙醇分别洗涤3 次和5 次,于60 ℃的真空烘箱中干燥24 h。最终样品命名为FS-VPQ。
1.3 Re(VII)的吸附实验
常温、常压、水溶液条件下,吸附剂对Re(VII)的吸附测试都是通过批次实验进行的。实验在7 mL与10 mL的聚乙烯离心管中,或是5 mL的一次性注射器管身中(接触时间极短时)进行。浓度为1 000 mg/L 的铼溶液由146.7 mg NaReO4于超纯水中溶解并定容至100 mL获得。溶液的pH由1~0.000 1 mol/L的HCl溶液稀释配制。单个吸附实验的溶液总体积为2 mL,分别由0.2 mL NaReO4溶液、0.2 mL HCl 溶液、0.2 mL 吸附剂分散液(固液比为4 mg/mL)和1.4 mL 超纯水组成,并根据具体实验类型调节某一变量。加料顺序为水、HCl溶液、铼溶液和吸附剂分散液,混合后将离心管置于脱色摇床中振荡(型号BETS-M5,海门市其林贝尔仪器制造有限公司)30 min,然后使用0.22 μm孔径的混合纤维素一次性针头过滤器进行固液分离。每批次实验进行时设置无吸附剂、其他因素一致的空白对照组,将其中铼的浓度作为初始浓度。Re(VII)的平衡吸附容量(qe)通过式(1)进行计算。

式中:c0为初始溶液中铼的浓度,mg/L;ce为吸附平衡时溶液中铼的浓度,mg/L;m为单个实验中使用的吸附剂质量,mg;V为单个实验中溶液体积,mL。
1.4 表征测试
F-SiO2微球的形貌由高分辨透射电子显微镜(HR-TEM,Hitachi H-7700,工作电压100 kV)和扫描电子显微镜(SEM,EVO 18,ZEISS)进行观察。氮气吸附与脱附等温线在温度77 K 下由比表面与孔隙度分析仪(TriStar II 3020,Micromeritics)表征完成。样品的比表面积通过多点BET 图表(Brunauer-Emmett-Teller)获得,总孔体积由P/P0=0.971时所有直径小于68 nm的孔的容积加和获得,孔径分布由BJH(Barrett-Joyner-Halenda)等温线的吸附分支通过微分获得。
样品表层的化学结构由X 射线光电子能谱仪(XPS,Thermo ESCALAB 250)表征获得,X射线源为单色Al-Kα(1 486.6 eV),N 1s 谱的通过能为30 eV,使用C 1s 284.8 eV 进行校准。样品的功能基团由傅里叶变换红外光谱(FTIR,Bruker TENSOR 27 FTIR)表征获得,样品与溴化钾按质量比1∶250 充分混合、研磨、压片后在波数4 000~400 cm-1内测量。样品的热重曲线(TGA)由热重分析仪(TGA Q5000)获得,初始温度为25 ℃,结束温度为800 ℃,升温速率为10 ℃/min,使用空气氛围。溶液中的金属离子浓度由电感耦合等离子发射仪(ICP-AES,Optima 7300DV,PerkinElmer,波长选取197.248 nm)进行测量。
2 结果与讨论
2.1 FS-VPQ的合成表征
FS-VPQ 的制备路线如图1 所示。首先使用硅烷偶联剂MPS 对F-SiO2进行双键改性,再将FSMPS 与溶剂甲醇、单体4-VP 和阻聚剂FeSO4混合后于γ 射线照射下进行辐射接枝,最后使用2-BETAB对FS-VP进行季铵化,得到产物FS-VPQ。
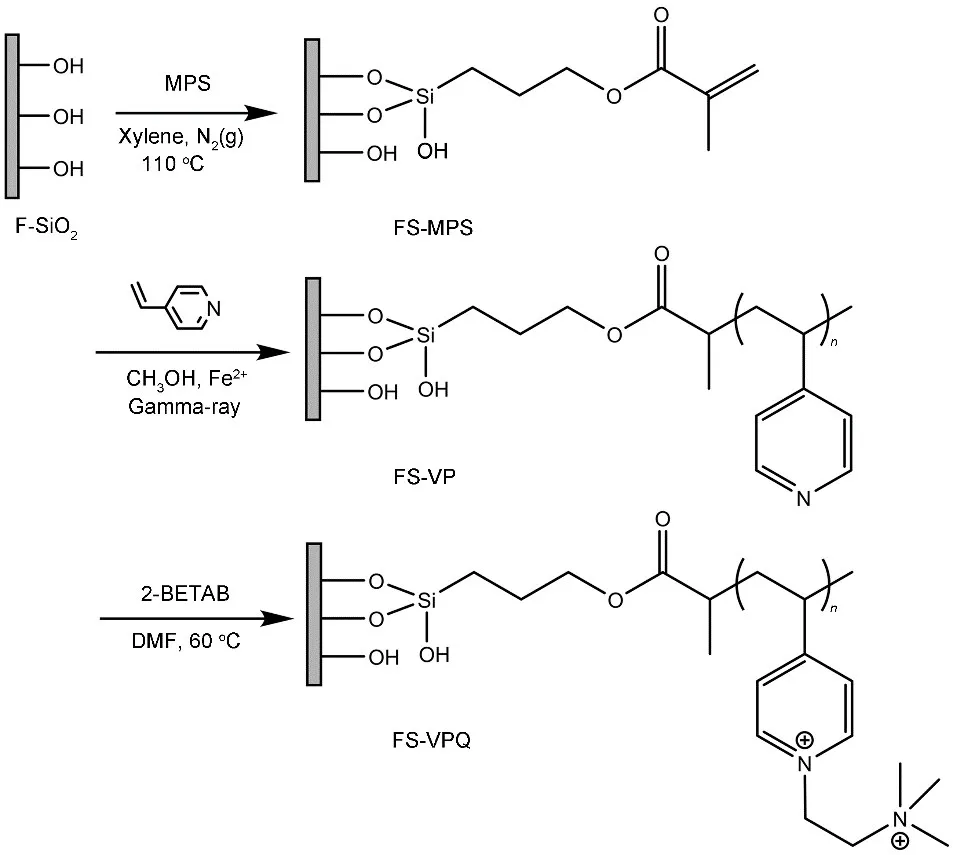
图1 从F-SiO2至FS-VPQ的合成路线图Fig.1 Synthesis schema of FS-VPQ from the modification of F-SiO2
F-SiO2与FS-VP 的傅里叶红外图谱如图2(a)所示。经吡啶功能化,FS-VP的图谱中出现了位于1 604 cm-1和804 cm-1(虽有重叠,但峰强显著增加)的吡啶环伸缩振动峰,位于557 cm-1的吡啶环弯曲振动峰和位于1 501 cm-1的环上C-H弯曲振动峰[25]。FS-VP 的XPS N 1s 图谱如图2(b)所示,吡啶中N原子的能级峰位于398.9 eV,同时因样品经过草酸溶液洗涤,存在一部分质子化吡啶,其中N原子的能级峰则位于400.9 eV。傅里叶红外和XPS数据说明成功合成了聚4-乙烯基吡啶功能化FSiO2。FS-VPQ 的XPS N 1s 图谱如图2(c)所示,除398.9 eV 的吡啶N 原子能级峰外,新增了位于402.0 eV 和400.7 eV 的两处能级峰。前者由2-BETAB 试剂中的季铵N 原子贡献,后者由季铵化步骤中的溶剂DMF 贡献,这说明成功合成了FSVPQ[26]。
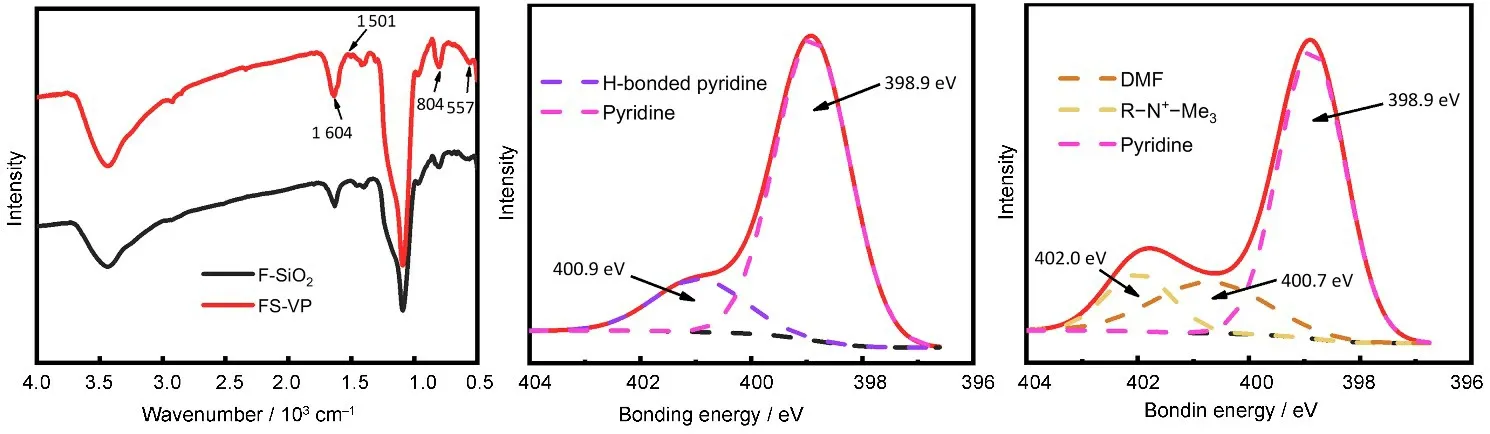
图2 F-SiO2和FS-VP的傅里叶红外图谱(a),FS-VP(b)和FS-VPQ(c)的XPS N 1s图谱Fig.2 FTIR spectra of F-SiO2 and FS-VP(a),XPS N 1s spectra of FS-VP(b)and FS-VPQ(c)
改性前后的的F-SiO2的TEM 和SEM 图像如图3所示。在改性前后样品都呈现出一致的近球形和内部放射状结构,F-SiO2与FS-VPQ 的平均半径分别为573 nm 和534 nm,说明制备F-SiO2纳米微球成功,并且改性和辐照过程不会对F-SiO2基体形貌造成显著的影响。FS-VPQ的TEM图像中,微球上出现明显的高分子聚合物,说明在改性过程中部分4-乙烯基吡啶发生了均聚。

图3 F-SiO2(a)和FS-VPQ(b)的TEM图像及FS-VPQ(c)的SEM图像Fig.3 TEM images of F-SiO2(a),FS-VPQ(b)and SEM image of FS-VPQ(c)
各样品的氮气吸附与脱附等温线和孔径分布图如图4所示。所有的样品均具有Type IV(a)型吸脱附等温线,并同时存在H4型滞后环,说明微球在季铵化改性过程中保留了较小的介孔[22]。而等温线在P/P0=0.7~0.9时的迅速爬升,说明微球内部同时存在较大的介孔。各样品的孔径分布则进一步证明微球具有不同孔径的多层次孔结构,其中孔径为3 nm 左右的介孔结构由F-SiO2的合成过程中双连续相微乳液造成,而位于12.5 nm附近的介孔结构则是微球中放射状纤维之间的间隙。样品的比表面积、总孔体积和介孔孔径大小如表1所示。随着改性的进行,材料的比表面积从F-SiO2的624.4 m2/g 逐步降至FS-VPQ 的333.6 m2/g,总孔体积从0.79 cm3/g逐步降至0.49 cm3/g,说明改性基团会占据孔内空间,且含有高分子结构的FS-VP 的比表面积和总孔体积下降幅度最大。
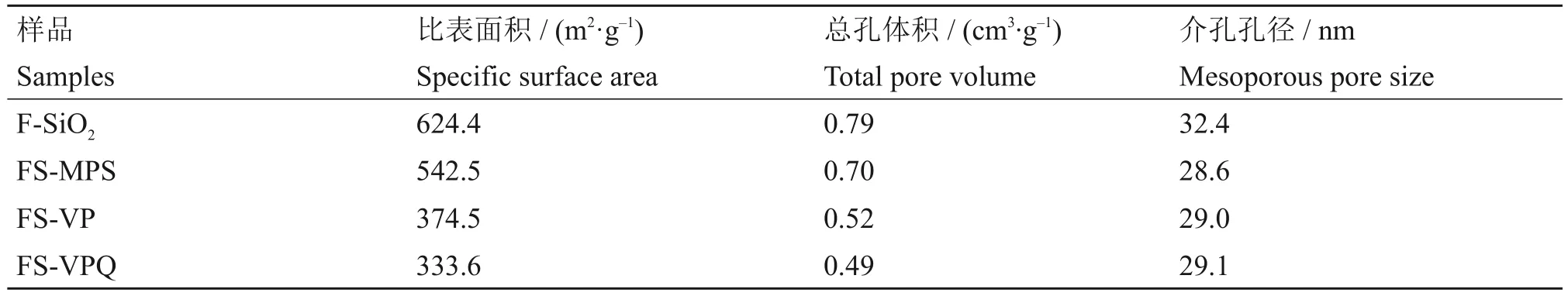
表1 F-SiO2及3种改性样品的比表面积、总孔体积和介孔孔径大小Table 1 Specific surface area,total pore volume and mesoporous pore size of F-SiO2 and three modified samples
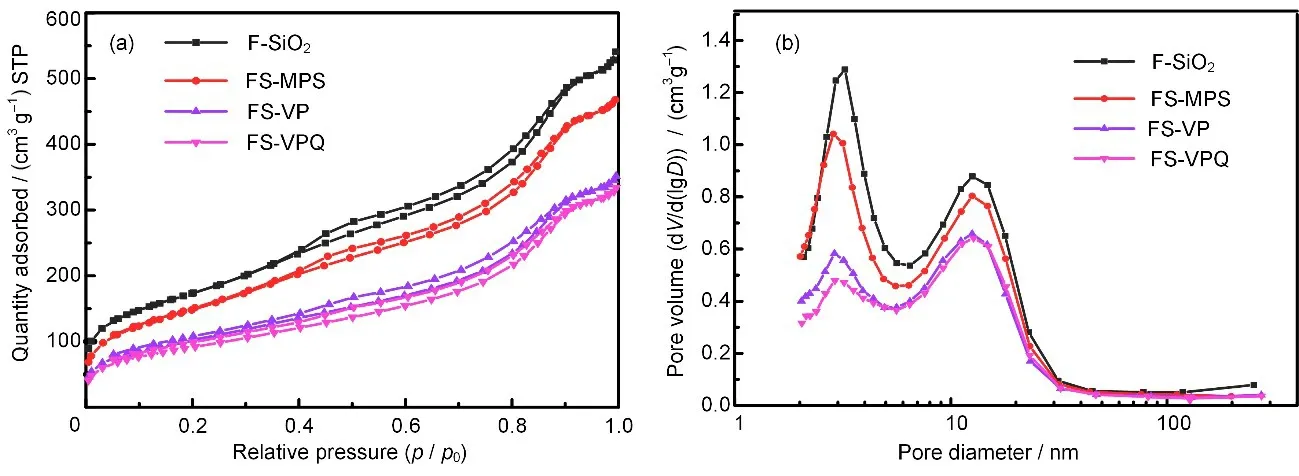
图4 F-SiO2及3种改性样品的氮气吸附与脱附等温线(a)和孔径分布图(b)Fig.4 N2 adsorption-desorption isotherms(a)and the corresponding pore size distributions(b)of different samples
样品的TGA 图谱如图5 所示。FS-VP 与FSVPQ在100 ℃以上时均出现明显失重现象,且FSVPQ的失重比例高于FS-VP,说明由F-SiO2微球表面改性的MPS,可通过辐射聚合于表面进一步接枝4-VP。
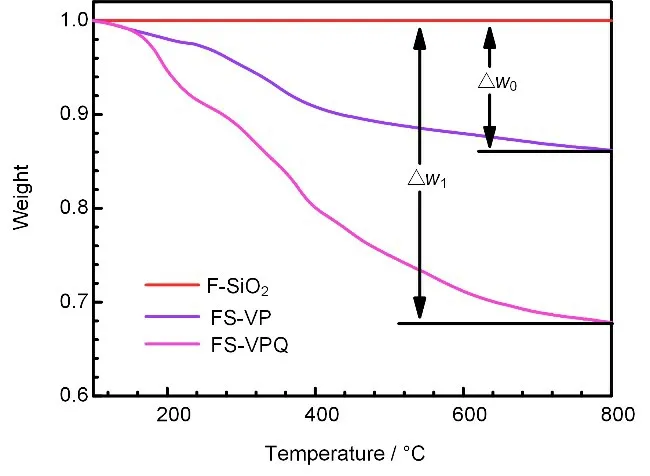
图5 F-SiO2、FS-VP和FS-VPQ的TGA图Fig.5 TGA figurs of F-SiO2、FS-VP和FS-VPQ
接枝率(RDG,%)通过两种样品相对于FSiO2基体的失重百分比Δw0和Δw1计算,计算方法见公式(2)。

2.2 FS-VP接枝率的影响因素研究
辐射接枝的接枝率越高,说明功能基团的数量越多,理论上对目标金属离子的吸附分离效果将会更好。通过优化辐射接枝步骤中的单体浓度、剂量率和总剂量,可以得到接枝率最高的产物。接枝率随各影响因素的变化见图6。
接枝率随单体浓度变化关系的曲线如图6(a)所示,FS-VP 的接枝率在单体浓度50%时有极大值。随着单体浓度的增加,接枝率越高。而单体浓度的增加使体系粘度上升,降低单体分子和FSiO2微球的扩散速率,接枝率下降,另外还有可能由于单体在FS-MPS 表层聚合速率过快形成紧密层,阻碍了后续接枝的进行[27]。
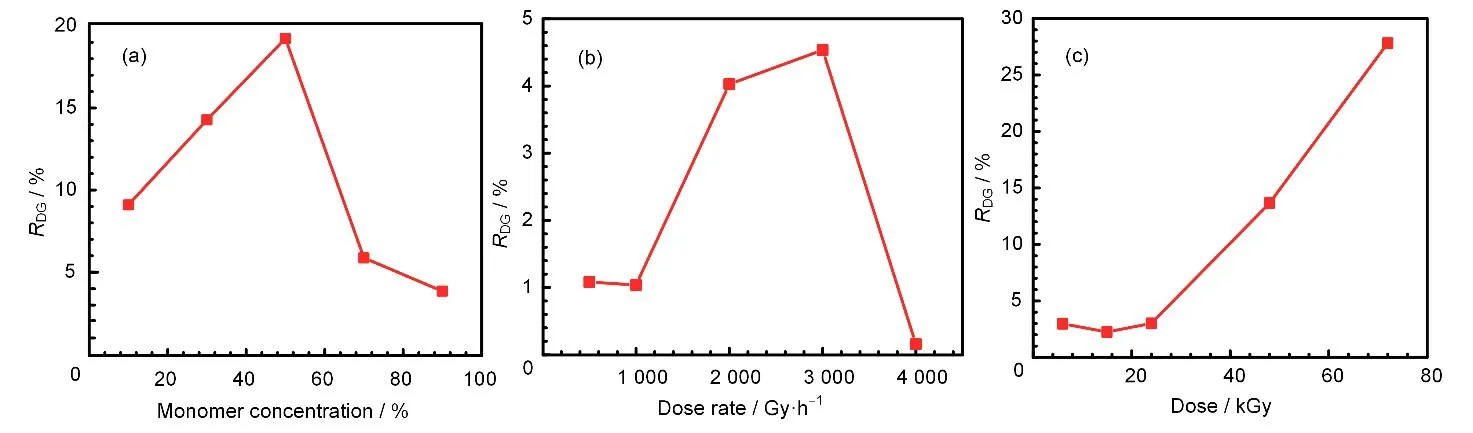
图6 4-VP的单体体积分数(a)、吸收剂量率(b)、吸收剂量(c)对FS-VP接枝率的影响Fig.6 Effect of monomer concentration of 4-VP(a),radiation dose rate(b),and absorbed dose(c)on grafting rate of FS-VP
接枝率随剂量率变化关系的曲线如图6(b)所示,FS-VP 的接枝率在剂量率为3 kGy/h 时有极大值。这是因为起初随着剂量率增大,辐射分解产生的初级粒子增加,使聚合反应的活性位点增多,促进接枝的进行,而剂量率过高时,则单体均聚现象加重,不利于接枝的进行[28]。
接枝率随吸收剂量变化关系的曲线如图6(c)所示,FS-VP 的接枝率在剂量小于24 kGy 时维持在较低水平,当剂量大于24 kGy 后则接枝率随着剂量的增加而增加。这是因为体系中自由基清除剂O2并未完全除尽,在其耗尽之前阻碍了接枝的进行。综上所述,当单体浓度为50%,剂量率为3 kGy/h,剂量为72 kGy 时,FS-VP 的辐射接枝率有极大值,为27.8%。后续实验中即使用接枝率为27.8%的FS-VP 进行季铵化并研究其对Re(VII)的吸附性能。
2.3 FS-VPQ对Re(VII)的吸附行为研究
2.3.1pH对吸附的影响
H+浓度是影响吸附剂对Re(VII)吸附性能的重要因素之一。样品在pH 为1~5 条件下,对Re(VII)的吸附容量如图7 所示,F-SiO2和FS-MPS因为没有吸附基团,其吸附容量极低。改性后FSVP和FS-VPQ的吸附容量则大幅提升,且FS-VPQ的吸附性能略高于FS-VP。FS-VP和FS-VPQ在pH为2 和3 时,吸附容量最高。随着pH 降低,Cl-与ReO4-竞争正电荷位点,ReO4-还可能与水合质子结合形成呈电中性的H9O4+·ReO4-粒子,使吸附容量降低。而pH升高时,吡啶质子化程度降低,正电荷被消除从而无法吸附ReO4-,使吸附容量降低。相比于FS-VP,FS-VPQ 基团中固有的两个正电荷使其具有更多的吸附位点,且在近中性下仍可吸附ReO4-,因此FS-VPQ 的吸附性能略高于FS-VP。以上因素使得吸附剂pH 为2 和3 时,对Re(VII)的吸附容量最高,为减少Cl-对吸附位点的竞争,后续实验选择pH为3时进行。
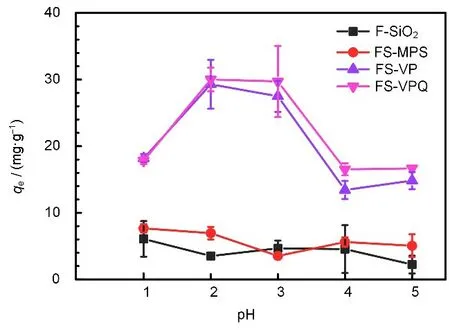
图7 F-SiO2及其3种改性样品在不同pH下对Re(VII)的吸附容量Fig.7 Adsorption capacity of F-SiO2 and its three modified samples for Re(VII)at different pH values
2.3.2吸附等温线
吸附等温线描述了溶质在吸附剂与溶剂的两相界面上吸附达到平衡时于两相中的分布情况。本实验在Re浓度为5~200 mg/L、溶液pH为3下使用FS-VPQ 进行吸附测试,并选取Langmuir 和Freundlich 两种常用吸附模型对数据进行拟合分析,它们可以分别用线性形式写成式(3)和(4)。
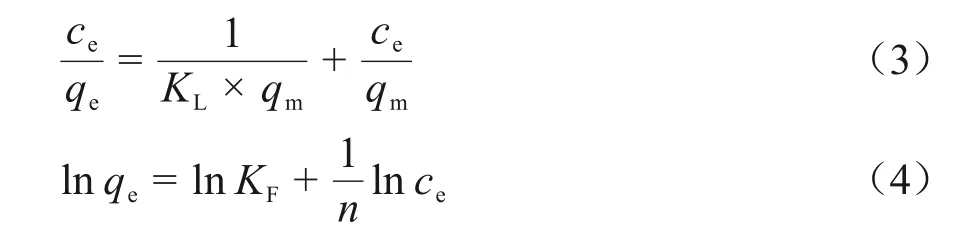
式中:qm是最大吸附量,mg/g;而KL是Langmuir吸附平衡常数[29],L/mg;KF是Freundlich 吸附平衡常数;n表示吸附强度。
经过拟合的图形和参数如图8所示。
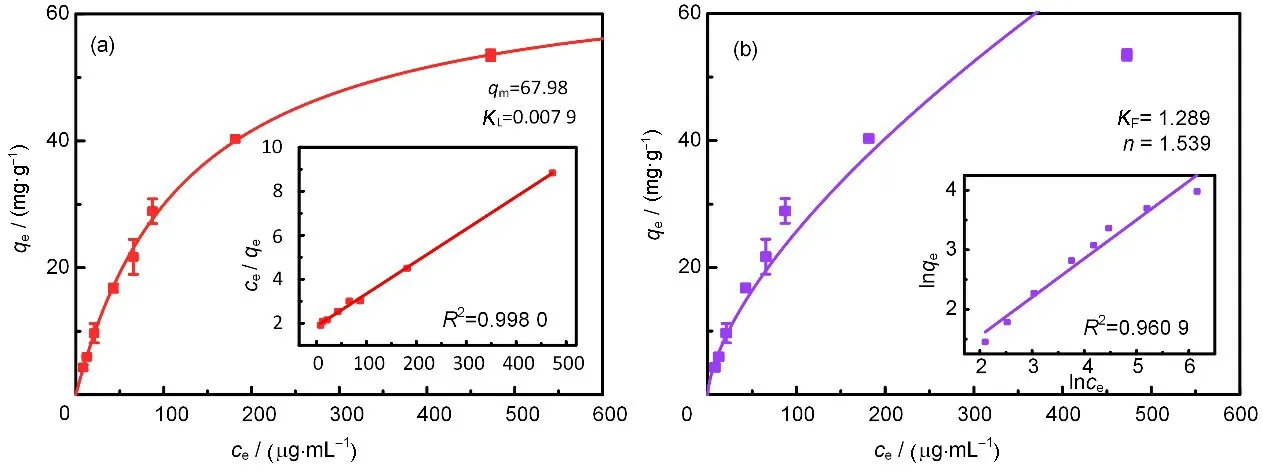
图8 FS-VPQ对Re(VII)的吸附等温线:Langmuir模型(a)和Freundlich模型(b)Fig.8 Adsorption isotherms of FS-VPQ to Re(VII):Langmuir model(a)and Freundlich model(b)
结果表明,使用Langmuir 模型拟合直线的R2(0.998 0)比使用Freundlich 模型拟合直线的R2(0.960 9)更接近1,并且图8(a)中的拟合曲线相比图8(b)中的拟合曲线更贴合实验值,说明FS-VPQ 对Re(VII)的吸附过程更适合用单分子层Langmuir 吸附模型来描述。由Langmuir 模型可得FS-VPQ 对Re(VII)的饱和吸附容量为68.0 mg/g,属于同类型吸附剂中较高性能,这源于FS-VPQ 中3 nm 左右的介孔带来的大比表面积和季铵化基团中固有的两个正电荷提供的吸附位点。
2.3.3吸附容量随时间的变化
在pH 为3 下对吸附容量随时间变化关系进行了研究,结果如图9 所示。吸附时间为3 min 时,吸附容量达到了24.8 mg/g,并且保持稳定,可以判断吸附于3 min内就已经达到平衡。较短的吸附平衡时间得益于FS-VPQ 中较大的介孔(12 nm 左右)结构带来的高传质效率,说明FS-VPQ在快速去除放射性污水中的Tc上有着推广应用的前景。
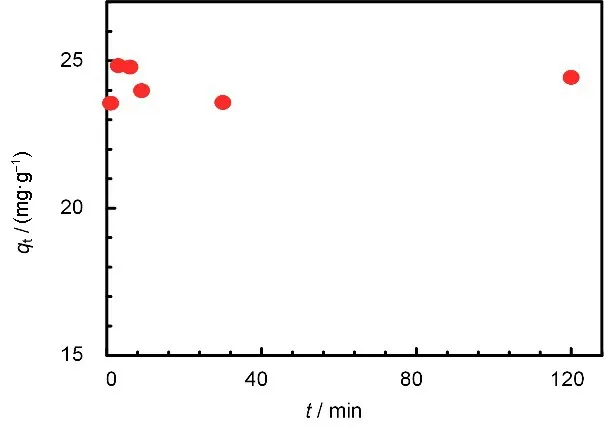
图9 FS-VPQ对Re(VII)的吸附容量随时间的变化Fig.9 Adsorption capacity of FS-VPQ to Re(VII)changes with time
2.3.4共存离子浓度对吸附的影响
在研究共存离子对Re(VII)的吸附影响中,总体积为2 mL 的吸附溶液由0.2 mL 铼溶液(1 000 mg/L)、0.2 mL 吸附剂分散液(固液比为4 mg/mL)、0.2 mL HCl 溶液(0.01 mol/L)以及一定体积的盐溶液(Na2SO4或NaCl 或NaNO3)和超纯水组成,其中Re 的初始浓度为100 mg/L(0.537 mmol/L),盐浓度为0.001~0.7 mol/L。不同浓度NaCl、Na2SO4和NaNO3对FS-VPQ 的吸附容量影响如图10所示。随着共存阴离子浓度的增加,Cl-、SO42-和NO3-会竞争正电荷位点,从而阻碍ReO4-的吸附,导致FS-VPQ对Re(VII)的吸附容量呈显著下降趋势。其中SO42-和ReO4-的几何构型类似,对吸附过程的影响程度大于Cl-和NO3-,导致SO42-存在时曲线的下降趋势更快。

图10 FS-VPQ在不同共存离子种类及浓度下对Re(VII)的吸附容量Fig.10 Adsorption capacity of FS-VPQ for Re(VII)under different types and concentrations of coexisting anions
3 结论
本文利用了辐射接枝法合成FS-VP 微球并对其进行季铵功能化改性,FTIR、XPS 和TGA 的数据结果共同验证了FS-VPQ的成功合成。在此基础上研究了单体浓度、剂量率和吸收剂量对产物接枝率的影响,得到在4-VP 单体体积分数50%、吸收剂量率3 kGy/h和剂量72 kGy的条件下FS-VP的接枝率有极大值27.8%。此外我们对FS-VPQ的Re(VII)吸附性能进行了研究,结果表明:季铵化改性带来的两个正电荷可提高吸附剂对Re 的吸附容量,并在近中性下仍有较好的吸附性能。FS-VPQ对Re(VII)的吸附等温线符合Langmuir模型,说明其为单分子层吸附。由于FS-VPQ具有多层次孔结构,较小介孔(3 nm)带来的高比表面积使其在pH为3时具有较大的吸附容量(68.0 mg/g);而较大的介孔带来的高传质速率使FS-VPQ 能够在3 min 内达到吸附平衡。共存阴离子对FS-VPQ 吸附性能的影响程度排序为SO42->NO3-≈Cl-。辐射改性方法简单,条件温和,所合成的FS-VPQ对Re具有较好的吸附性能,在去除放射性污水中的Tc方面具有潜在的应用价值。
作者贡献说明林铭章和翁汉钦提出了本文的研究思路和实验方案。沈旺来完成了本工作中所有介孔SiO2材料的改性合成、表征以及对铼的吸附实验。陈怡志和沈旺来共同完成了本论文初稿的撰写。张鹏对本工作中得到的实验数据进行了处理。吴志豪对本文进行了校对和语言修改。所有作者均已阅读并认可该论文最终版的所有内容。
猜你喜欢
分子催化(2022年1期)2022-11-02
天津大学学报(自然科学与工程技术版)(2022年11期)2022-11-01
建材发展导向(2022年1期)2022-03-08
建材发展导向(2021年19期)2021-12-06
安全与环境工程(2021年5期)2021-10-08
发明与创新·小学生(2020年4期)2020-08-14
科学导报·学术(2019年19期)2019-09-10
分析化学(2018年8期)2018-11-01
分析化学(2016年7期)2016-12-08
发明与创新·小学生(2016年4期)2016-08-04
