
溴代蒽并及酚嗪并噻二唑衍生物的合成及光电性能研究
2022-03-04 07:46李雅琴吴进军刘茂松樊明轩余响林黎俊波
武汉工程大学学报 2022年1期
李雅琴,吴进军,刘茂松,樊明轩,余响林,黎俊波*
1.武汉工程大学化学与环境工程学院,湖北 武汉 430205;2.武汉工程大学化工与制药学院,湖北 武汉 430205
多并苯材料是一类苯环线性组合的有机半导体材料[1-3],目前已经在有机半导体领域如有机场效应晶体管[4-5]、有机太阳能电池[6-7]、有机发光二级管[8-9]、有机存储器件[10-11]等多个领域得到广泛应用。多并苯材料作为有机半导体器件活性组分一个明显的缺点就是稳定性较差,而杂原子掺杂,特别是氮原子掺杂构建的氮杂多并苯不但能提高多并苯材料的稳定性[12],还能有效调控多并苯材料电子传输性能。
目前,许多构建氮杂多并苯材料合成子已经被报道,其中蒽或酚嗪并噻二唑为一类构建吡嗪类特别是具有较大骨架吡嗪类氮杂多并苯材料的常用合成子[13],对于新型氮杂多并苯骨架构建主要基于改变邻二碳基合成子结构从而有效扩充氮杂多并苯材料结构[14-15],目前大量二羰基化合物结构已被报道,同时,二羰基化合物相对来说结构稳定性较差,提纯难度大,进一步拓展二羰基化合物结构受到限制。基于此,我们设想通过拓展蒽或酚嗪并噻二唑骨架结构,为氮杂多并苯材料结构拓展提供空间。通过在蒽或酚嗪并噻二唑结构的6,7位上修饰溴原子,制备溴代蒽或酚嗪并噻二唑,在合成子中引入活性溴原子,为其进一步结构修饰提供可能位点,有望大大扩展吡嗪类氮杂多并苯材料结构。
本文巧妙地分别利用含溴取代的活化卤化物与苯并噻二唑-4,7-二酮发生Diels-Alder反应[16-18],合成了不同溴代位点的蒽并噻二唑氮杂多并苯衍生物。同时,为了构建更多的噻二唑氮杂并苯分子,选择从二胺类砌块出发,通过分别与溴代芳香二酮化合物进行分子内缩合反应,合成了不同溴代位点的酚嗪噻二唑氮杂多并苯衍生物。通过这两种合成方法,成功地在蒽并及酚嗪噻二唑骨架上的6,7位引入溴原子,并对溴原子位置对材料的光电性能的影响进行了探讨,该分子设计理念为氮杂并苯材料结构拓展提供了新思路。
1 实验部分
1.1 仪器与试剂
仪器:核磁共振仪(美国瓦里安公司VX 400);BiflexⅢ型质谱仪(Bruker公司)紫外分光光度计(日本岛津公司);荧光光谱仪(日本日立公司F-7000);电热恒温鼓风干燥箱(DHG-9140A);电子天平(FA2204B)。
试剂:对苯二甲醚、醋酸钾、冰乙酸、乙酸乙酯、二氯甲烷、无水三氯化铝、无水硫酸钠、氧化银、1,4-二 氧 六 环、N-溴 代 琥 珀 酰 亚 胺(NBromosuccinimide,NBS)、盐酸、N,N-二甲基甲酰胺(N,N-Dimethylformamide,DMF)、三异丙基硅基乙炔、四氢呋喃、正丁基锂、碘化钾、次亚磷酸钠、2-溴-6-甲氧基苯酚、2,3-二氯-1,4萘醌、4-溴-1,2-二甲苯、四丁基溴化胺、4-溴邻苯二酚、苯并噻二唑、三丁基氯化锡、双三苯基磷二氯化钯铵均为市售分析纯,且所有试剂及溶剂均经过常规纯化处理,用水为二次蒸馏水。
1.2 实验方法
1.2.1 设计合成的4种化合物 4种化合物的结构式如图1所示。
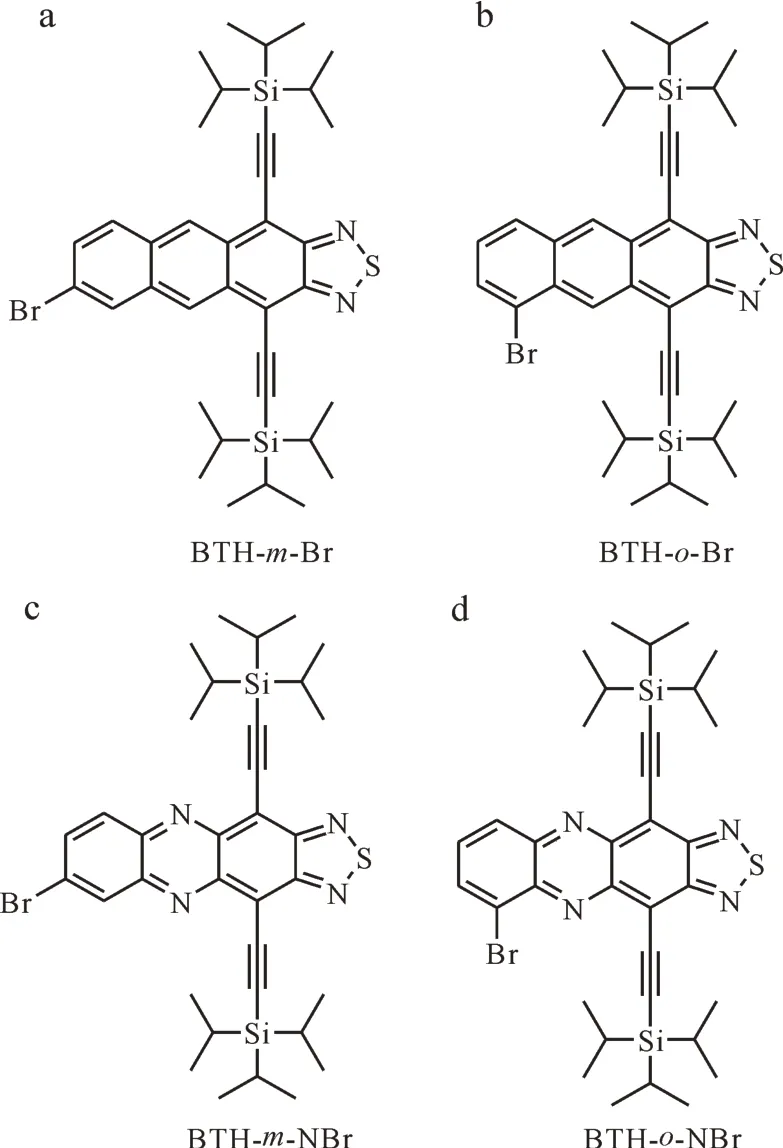
图1 设计合成的4种化合物:(a)BTH-m-Br,(b)BTH-o-Br,(c)BTH-m-NBr,(d)BTH-o-NBr Fig.1 Designed and synthesized four compounds:(a)BTH-m-Br,(b)BTH-o-Br,(c)BTH-m-NBr,(d)BTH-o-NBr
1.2.2 合成路线 4种化合物的合成路线如图2所示。

图2 4种化合物的合成路线Fig.2 Synthetic route of four compounds
1.3 实验步骤
1.3.1 化合物3a、3b的合成[19]室温下将化合物1(0.69 g,4.2 mmol),化合物2a(2.40 g,4.8 mmol)和碘化钾(2.76 g,16.8 mmol)加入到100 mL的两口烧瓶中,加入40 mL的无水DMF,反应抽真空通入氮气保护,将混合物加热至110℃反应24 h,反应结束后减压蒸馏除去溶剂,得到棕褐色油状液体,通过硅胶色谱柱法纯化(洗脱剂:二氯甲烷)得到黄色产物(0.216 g,产率16%)。表征与已合成化合物一致,证明为化合物3a。将化合物2a更换成2b,合成步骤同3a的合成,制备得化合物3b黄色产物(0.22 g,产率23%)
1.3.2 化合物6a、6b的合成 将8 mmol的化合物5a四溴邻苯二酚溶于30 mL的二氯甲烷中,同时加入5~10 mg四丁基溴化胺溶解。加入NaIO4(2 g,9.3 mmol)溶于20 mL二氯甲烷溶液,将混合溶液在室温下搅拌20 min以上,同时TLC监测。待反应结束将各层分离,合并有机相用无水硫酸钠干燥,蒸发溶剂得红棕色固体6a。将化合物5a更换成5b,合成步骤同6a的合成,制备得化合物6b(橙色固体)。因其二酮化合物不稳定,直接将新制的化合物6a、6b投入下一步反应。
1.3.3 化合物BTH-m-Br的合成[19]在0℃的条件下,将100 mL的两口烧瓶置于冰浴的环境中,反应装置抽真空,通入氮气保护,随后向反瓶中注射20 mL干燥的THF溶液,在搅拌过程中依次注射三异丙基硅基乙炔(0.9 mL,3.90 mmol)和1.6 mol/L的正丁基锂溶液(1.5 mL,3.0 mmol),将混合物置于室温环境下搅拌0.5 h。然后再将化合物3a(0.27 g,0.78 mmol)加入到溶液中并搅拌过夜,待反应结束后加入1 mL无水乙醚淬灭。蒸发溶剂后通过柱层析法(洗脱剂:甲醇)纯化。合并产物加入到20 mL的乙酸中,依次加入次亚磷酸钠(0.78 g,7.5 mmol)和碘化钾(1.29 g,7.71 mmol)将混合物回流1 h,待反应结束冷却至室温,加入100 mL水,再用二氯甲烷萃取,合并有机相用无水硫酸钠干燥。蒸发溶剂得到黑蓝色粉末,用乙醇洗涤抽滤得产物BTH-m-Br(368 mg,产率70%)。
1.3.4 化合物BTH-o-Br由类似BTH-m-Br的方法合成 将化合物3a更换为3b,合成步骤同BTHm-Br的合成。最终得到BTH-o-Br(279 mg,产率79%)。1H NMR(500 MHz,Chloroform-d)δ9.62(s,1H),9.23(s,1H),7.89(d,J=8.6 Hz,1H),7.74(d,J=6.9 Hz,1H),7.23(d,J=7.8 Hz,1H),1.34-1.31(m,42H)。13CNMR(126 MHz,CDCl3)δ153.32,153.19,133.47,132.88,132.73,131.44,130.51,128.70,127.73,127.46,126.54,123.22,113.32,112.20,109.26,102.22,18.96,18.89,18.45,11.51.MS(MALDI)m/z Calc.for[M+H]+C36H47BrN2SSi2:676.2,found:677.0。
1.3.5 化合物BTH-m-NBr的合成 在干燥的两口烧瓶中加入化合物7(280 mg,0.531 mmol)和化合物6a(294 mg,1.57 mmol),反应抽真空,并通入氮气保护。注射加入8 mL冰乙酸和8 mL二氯甲烷,混合物在室温下搅拌17 h。用饱和碳酸氢钠溶液处理反应体系中的乙酸并加入二氯甲烷萃取,用无水硫酸钠干燥有机相层并抽滤。蒸发溶剂通过柱层析(洗脱剂为石油醚/二氯甲烷,体积比5∶1)得蓝黑色晶体BTH-m-NBr(245 mg,产率:68%)。1H NMR(400 MHz,Chloroform-d)δ8.33(s,1H),7.99(d,J=9.4Hz,1H),7.82(d,J=9.4 Hz,1H),1.31~1.25(m,42H)。13C NMR(126 MHz,Chloroform-d)δ154.67,145.23,143.97,142.60,142.45,135.98,132.07,131.52,127.39,114.68,112.34,101.91,18.90,18.89,11.56.MS m/z Calc.for[M+H]+C34H45N4BrSSi2:678.90,found:678.15。
1.3.6 化合物BTH-o-NBr由类似BTH-m-NBr的方法合成 将化合物6a更换为6b,合成步骤同BTH-m-NBr的合成。最终得BTH-o-NBr(34 mg,0.05 mmol,产率:18.8%)。1H NMR(400 MHz,Chloroform-d)δ8.16(dd,J=7.1 Hz,1.2 Hz,1H),8.11(dd,J=8.9 Hz,1.2Hz,1H),7.64(dd,J=9.0,7.1Hz,1H),1.36-1.25(m,42H)。13CNMR(126MHz,Chloroform-d)δ155.34,154.88,145.61,142.50,135.14,132.04,130.26,125.15,115.10,112.70,112.34,101.95,18.95,18.90,11.57。MS m/z Calc.for[M+H]+C34H45N4BrSSi2:678.90,found:678.15。
2 结果与讨论
2.1 合成方法
化合物1以对甲醚为起始原料由文献[20]方法经过五步反应合成得到,总收率(57%),化合物2以4-溴-1,2-二甲苯为原料通过NBS自由基反应得到[19],总收率(50%)。狄尔斯-阿尔德反应(Diels-Alder反应),作为有机合成中一种构建碳碳键重要的手段,因其高效简洁的合成步骤,常被用作构建更大骨架氮杂并苯。本文通过将噻二唑并苯醌与四溴邻二甲苯发生原位D-A缩合反应得到化合物3,利用TIPS与羰基亲核加成然后消除反应得到不同溴取代的蒽并噻二唑化合物。缩合反应(尤其是二胺与二酮)与偶联反应是制备氮杂蒽最常用的方法,由于二胺(或四胺)通常在结构上更容易调节,本文中,通过采用从二胺砌块出发,通过分别与3,4位溴取代的芳香二酮化合物进行分子内缩合反应,合成了不同溴代位点的酚嗪噻二唑氮杂多并苯衍生物。而其中4-溴邻苯二醌的稳定性明显高于3-溴邻苯二醌,进一步缩合得到酚嗪并噻二唑时合成BTH-m-NBr产率(68%)明显高于BTH-o-NBr(19%)
2.2 光学性质
如图3(a)中显示了化合物BTH-m-Br,BTH-o-Br,BTH-m-NBr,BTH-o-NBr的 紫 外 吸 收 光 谱(10-5mol/L,CH2Cl2)。在450~650 nm之间所有化合物的紫外吸收展现出2组吸收峰,此阶段可以归因于分子内的电荷转移。化合物BTH-m-Br,BTH-o-Br,BTH-m-NBr,BTH-o-NBr显示出近似的吸收峰。酚嗪并噻二唑化合物BTH-m-NBr相比蒽并噻二唑化合物BTH-m-Br蓝移11 nm左右,酚嗪并噻二唑化合物BTH-o-NBr和蒽并噻二唑化合物BTH-o-Br相比蓝移9 nm左右,间溴类化合物BTH-m-NBr相对邻位溴代类化合物BTH-o-NBr蓝移2 nm。间溴类化合物BTH-m-Br相对邻位溴代类化合物BTH-o-Br蓝移1 nm。
4种化合物的起始吸收峰分别为686、680、683、688 nm。根据公式Egap=1 240/λonset计算出4种化合物的光学带隙分别为1.81、1.82、1.82、1.80 eV(其中溴代的蒽/酚嗪并噻二唑结构与文献[21]报道的不含取代基的TIPS基蒽并噻二唑化合物起始吸收664 nm的光学带隙为1.87 eV相比较小)。由图3(b)显示出4种化合物BTH-m-Br、BTH-o-Br、BTH-m-NBr、BTH-o-NBr的发射峰分别位于699、702、685、687 nm。对比蒽并噻二唑化合物BTH-m-Br和酚嗪并噻二唑化合物BTH-m-NBr的荧光发射,发现氮原子掺杂到分子的骨架中有轻微增强荧光的作用,这可能是氮原子掺杂部分阻断了分子的共轭体系造成的。
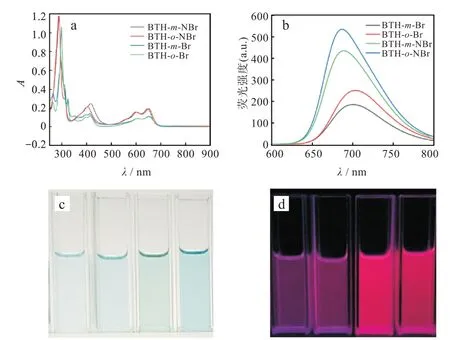
图3 4种化合物的光学性质:(a)紫外吸收光谱,(b)荧光光谱,(c)可见光,(d)荧光Fig.3 Optical properties of four compounds:(a)ultraviolet absorption spectra,(b)fluorescence spectra,(c)visible light,(d)fluorescence
在实验过程中以9,10-二苯基蒽的CH2Cl2标准参比液(Φ1=0.95)测试4种化合物的荧光量子产率。利用荧光量子产率计算公式将测得的参数代入进行荧光量子产率的计算,计算公式为:
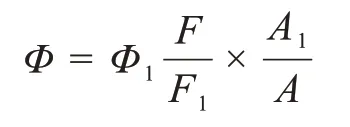
其中Φ、Φ1分别为待测物和标准物的荧光量子产率,F为待测物、F1为标准物的荧光积分强度,A、A1分别为在激发波长下待测物和标准物的最大吸光度。通过测试化合物BTH-m-Br、BTH-o-Br、BTH-m-NBr、BTH-o-NBr所得的荧光量子产率计算出结果分别为0.022、0.029、0.056、0.053。
研究结果表明氮原子个数及溴取代位置对光电化学性质影响较大,氮原子引入使最大吸收波长分别蓝移11、9 nm;溴原子位置对其光学性质的影响可忽略不计;其中溴代苯并酚嗪的荧光最强,溴代酚嗪并噻二唑的荧光稍强于溴代蒽并噻唑。
2.3 电化学性质
采用循环伏安法在无水二氯甲烷溶液中测试了 化 合 物BTH-m-Br、BTH-o-Br、BTH-m-NBr、BTH-o-NBr的电化学性质(扫描速率为100 mV/s)。使用0.1 mol/L四丁基六氟磷酸铵(Bu4NPF6)作为电解质,二茂铁作为标定物,玻碳为工作电极,铂丝为参比电极和支撑电极。图4给出了4种化合物的循环伏安曲线。从图可知化合物BTH-m-Br和BTH-o-Br显示3组可逆的氧化和还原峰及1个不可逆的氧化峰。而化合物BTH-m-NBr、BTH-o-NBr显示出2组可逆的还原峰和2个不可逆的氧化峰。4种化合物的起始氧化电位分别为0.72、0.70、0.97、0.94 V。表明蒽并噻二唑类化合物较酚嗪并噻二唑类化合物易氧化。通过方程式计算EHOMO=-(4.8+Eonsetox)。其中化合物BTH-m-Br、BTH-o-Br、BTH-m-NBr、BTH-o-NBr的HOMO能级计算为-5.52,-5.50,-5.77,-5.74 eV。同时,化合物的起始还原电位分别为-1.03,-1.06,-0.75,-0.72 V,说明氮掺杂的酚嗪噻二唑化合物易被还原,可归因于碳骨架中掺杂氮原子破坏分子共轭体系造成的。通过氧化还原电位差方程式ELUMO=-(4.8+Eonsetred)计 算 最 低 未 占 据 轨 道(lowest unoccupied molecular orbital,LUMO)能 级 分 别为-3.77、-3.74、-4.05、-4.08 eV。氮原子引入能够降低同结构化合物的LUMO能级,同时电化学起始氧化电位升高0.22~0.27 V;而溴原子位置的不同对其氧化还原峰无影响。本实验采用了Gaussian09(以DFT/B3LYP/6-31G为基组)的方式对已合成化合物中的BTH-m-Br/o-Br的LUMO和最高占据轨道(highest occupied mollcule orbital,HOMO)电子云分布进行了理论模拟计算,结果如表1所示。同时将实际实验数据和理论计算数据汇总于表1中。结合理论计算和实际实验测试结果分析,4种化合物的LUMO和HOMO能级及能带的变化与理论计算结果趋于一致。同时通过理论计算,图5给出了4种化合物的能级电子云分布。
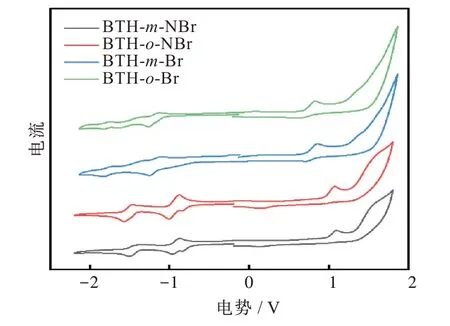
图4 四种化合物的循环伏安曲线Fig.4 Cyclic voltammetry curvesof four compounds

图5 化合物能级电子云分布:(a)BTH-m-Br,(b)BTH-o-Br,(c)BTH-m-NBr和(d)BTH-o-NBr的LUMO和HOMOFig.5 Electron cloud distribution of LUMO and HOMO energy levels of compounds:(a)BTH-m--Br,(b)BTH-o-Br,(c)BTH-m--NBr and(d)BTH-o-NBr
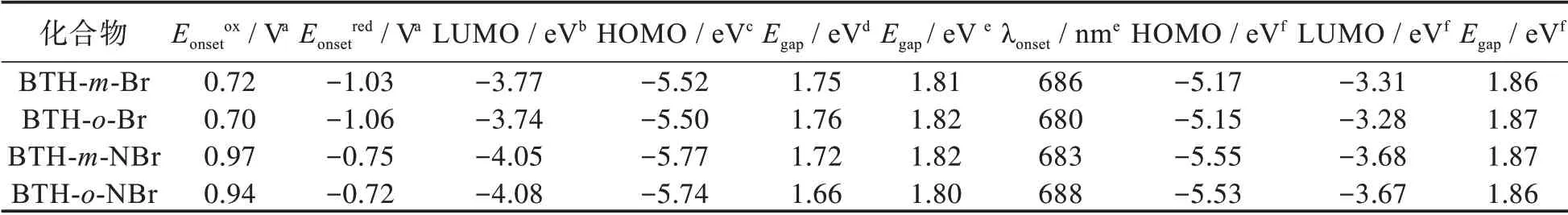
表1 4种化合物的光学和电化学数据Tab.1 Data of photoelectrical properties of four compounds
3 结 论
本文通过将噻二唑并苯醌与四溴邻二甲苯发生原位D-A缩合反应,再利用TIPS与羰基亲核加成然后消除反应得到不同溴取代的蒽并噻二唑化合物。同时通过采用从二胺砌块出发,通过二胺砌块分别与3,4位溴取代的芳香二酮化合物进行分子内缩合反应,合成了不同溴代位点的酚嗪噻二唑氮杂多并苯衍生物,丰富了噻二唑类合成子种类,为新型氮杂多并苯结构拓展提供了新的思路。后续研究将利用溴代蒽并噻二唑及酚嗪并噻二唑合成子构建不同功能的氮杂多并苯材料。
猜你喜欢
可再生能源(2022年5期)2022-06-09
城市道桥与防洪(2022年3期)2022-05-08
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
安全与环境工程(2021年2期)2021-04-02
今日农药(2017年10期)2017-11-14
科技与创新(2015年20期)2015-10-29
科技与企业(2015年20期)2015-10-21
