
氧化石墨烯基钴卟啉复合材料的电催化析氢反应
2022-03-01 06:48:02谌小燕陈卫昌肖新颜刘海洋
高等学校化学学报 2022年2期
俞 彬,谌小燕,赵 越,陈卫昌,肖新颜,刘海洋
(华南理工大学化学与化工学院,广东省燃料电池技术重点实验室,广州 510641)
氢气是可再生、热值大及便于利用的清洁能源.电解水制氢是氢气产生的重要途径[1].铂是目前电解水制氢最优异的催化剂[2],但由于其在自然界中含量很低、价格十分昂贵,驱使人们寻找其它自然界含量丰富的铁[3]、钴[4]、镍[5]等过渡金属催化剂.过渡金属卟啉配合物因具有多种优点而引人注目[6]:(1)卟啉环平面刚性结构和合适的空腔可为中心金属提供稳定的配位环境,可结合多种过渡金属和主族元素;(2)卟啉环周边容易修饰上不同的功能基团,可以组装或接枝在材料上;(3)作为电产氢催化剂,金属卟啉容易形成低价金属-氢中间体.目前,卟啉电催化制氢催化剂主要是用于均相体系,催化剂分子结构设计通常在于改变卟啉配合物的中心金属[7,8]、中位置取代基[9]、轴向配体[10]或使用聚卟啉[11]等;如Beyene等[12]研究表明,苯磺酸基的水溶性铜(II)卟啉在中性水溶液中电催化制氢的电位为−470 mV,法拉第效率达到98.3%.以叔胺作为助催化剂时,利用四苯基卟啉铁[13]以乙酸为质子源电催化产氢时,乙酸和胺的加合物可使质子活性增强,显著提高产氢量.
石墨烯是一种二维碳纳米材料,具有优良的导热性、导电性和高的机械强度,但石墨烯层与层之间存在强的范德华力,使其在溶液中容易发生聚集,因此其分散性能较差[14].氧化石墨烯(GO)的表面经过含氧官能团的修饰,可以较好解决分散性差的问题,而且其表面含氧官能团通常也作为其它催化剂的结合位点.将具有电子受体能力的GO与具有电子供体性质的卟啉结合,通过GO与卟啉之间的高电子传输效率提高材料的电催化性能[15].卟啉石墨烯基材料的制备包含共价键和非共价键两种方法.共价键结合材料主要是利用卟啉与石墨烯之间的酯键、酰胺键等连接[16],而非共价键结合材料则利用卟啉与石墨烯之间的范德华力、氢键、π⁃π堆积与静电作用作为组装的驱动力[17].利用GO与金属卟啉的π⁃π堆积作用来构筑卟啉石墨烯基材料是一种重要的方法[18~20].
金属卟啉与GO材料结合后可以降低其电催化析氢的起始过电位.目前,大多报道的共价结合卟啉石墨烯材料主要用于光催化制氢[21],利用共价结合的卟啉石墨烯做电催化析氢材料的研究很少,仅见最近Zhu等[22]利用酰胺键将聚四苯基卟啉(TPPCOP)锚定在石墨烯上制得了GO-TPPCOP材料,其电流密度10 mA/cm2对应的过电位为−376 mV.而在卟啉石墨烯电催化析氢材料方面,则主要利用非共价结合来构筑.2014年,Wang等[23]首次采用静电作用将带正电荷的四(N-甲基吡啶基)卟啉钴(CoTMPyP)与GO自组装并经过电化学还原(ER)制得了[ERGO@CoTMPyP]n多层薄膜材料,其中[ERGO@CoTMPyP]7材料的电流密度1 mA/cm2对应的过电位为−474 mV.Ma等[24]采用同样的方法将CoTMPyP负载在纳米碳片上制备了CoTMPyP/ERGO材料,其析氢的起始电位为−220 mV,电流密度1 mA/cm2对应的过电位为−315 mV.2017年,Lee等[25]通过在二维单层石墨烯固液界面直接合成了四(对-氨基苯基)卟啉(TAPP),制备了M(Zn,Ni,Pt)-TAPP/G复合材料,Pt-TAPP/G的电催化析氢起始电位降低为−160 mV,电流密度1 mA/cm2对应的过电位为−175 mV.最近,Chen等[26]通过钴酞菁与氧化石墨烯π⁃π堆积制备的PyCoPc/GO复合材料在电流密度10 mA/cm2时的过电位为−373 mV,Tafel斜率为145 mV/dec;进一步高温碳化得到PyCoPc/GO-800材料在电流密度10 mA/cm2的过电位上升为−253 mV,Tafel斜率下降为96 mV/dec.基于以上研究,本文直接利用5,10,15,20-四(苯基)卟啉钴(CoTPP)与氧化石墨烯的π-π堆积作用构筑了CoTPP/GO复合材料,结果发现,该材料具有良好的电催化析氢性能,且CoTPP与GO的质量比为1∶15时,材料的性能最好.向体系中加入二苯硫醚(SPh2)轴向配体后,CoTPP(SPh2)/GO的析氢起始电位达到了−235 mV,其性能接近目前最好的卟啉/石墨烯Pt-TAPP/G电催化析氢材料.
1 实验部分
1.1 试剂与仪器
吡咯(C4H5N,纯度≥98%)、苯甲醛(C6H5CHO,纯度≥98%)、丙酸(CH3CH2COOH,纯度≥98%)、四水合乙酸钴[Co(OAc)2·4H2O,纯度≥99.5%]、二氯甲烷(DCM,纯度≥99.5%)、二苯硫醚(SPh2,纯度≥98%)、全氟磺酸-聚四氟乙烯共聚物(Nafion,纯度≥98%)、无水乙醇(CH3CH2OH,纯度≥99.5%)、N,N-二甲基甲酰胺(DMF,纯度≥99.5%)、氧化石墨烯(GO,纯度≥99.5%)、盐酸(HCl,纯度≥99.5%)和氯化钠(NaCl,纯度≥99.5%)均购自国药集团化学试剂有限公司.饱和食盐水需要用氯化钠现配现用.
Hitachi U-3010型紫外-可见光谱仪(UV-Vis)和Hitachi U-3010型紫外-可见漫反射光谱仪(UV-Vis DRS)(日本Hitachi公司);Bruker Avance型核磁共振波谱仪(NMR)和Bruker maxis impact型高分辨质谱联用仪(HRMS,德国Bruker公司);ZEISS MERLIN Compact扫描电子显微镜仪(SEM,德国ZEISS公司);HJY LabRAM Aramis型共聚焦显微拉曼光谱仪(Raman),法国H.J.Y公司;Thermo Scientific K-Alpha+型X射线光电子能谱仪(XPS,美国Thermo Fisher Scientific公司);CHI-660E电化学工作站(上海辰华仪器有限公司);Agilent Technology 7890A气相色谱仪(GC,美国Agilent Technologies公司).
1.2 实验方法
1.2.1 5,10,15,20-四(苯基)卟啉的制备 参照文献[27]的方法,将67.09 mg(1 mol)吡咯和106.12 mg(1 mol)苯甲醛加入100 mL的圆底烧瓶中,再加入30 mL丙酸溶剂,并置于油浴锅中加热搅伴使温度升至135℃,反应2 h后停止加热和搅拌.反应结束后待恢复至室温,滤除反应液,用水洗涤3~4次,再用甲醇洗涤3~4次,烘干溶剂得到紫色粉末状固体5,10,15,20-四(苯基)卟啉(TPP)95.22 mg,产率为62%.
1.2.2 5,10,15,20-四(苯基)卟啉钴的制备 参照文献[28]的方法,将61.43 mg(0.1 mmol)TPP和24.91 mg(1.4 mmol)Co(OAc)2·4H2O加入100 mL的圆底烧瓶中,并加入30 mL DMF溶剂,置于140℃油浴锅中搅拌下进行反应,3 h后停止加热.待反应液冷却至室温后,加入100 mL DCM,用饱和食盐水洗涤4~5次,收集有机相,旋干溶剂后,用柱层析分离纯化.最终得到褐色粉末状固体5,10,15,20-四(苯基)卟啉钴(CoTPP)58.39 mg,产率为87%.
1.2.3 5,10,15,20-四(苯基)卟啉钴(二苯硫醚)的制备 参照文献[29]的方法,将67.12 mg(0.1 mmol)CoTPP和223.52 mg(1.2 mmol)SPh2加入100 mL的圆底烧瓶中,并加入30 mL DCM,置于油浴锅中加热回流反应3 h.待冷却至室温后,过滤得到黑色反应固体即为最终粗产物,进一步重结晶提纯,将得到的固体产物置于真空干燥箱中干燥,最终得到黑色粉末状固体5,10,15,20-四(苯基)卟啉钴(二苯硫醚)[CoTPP(SPh2)]72.09 mg,产率为84%.
1.2.4 5,10,15,20-四(苯基)卟啉钴/GO电催化剂的制备 参照文献[30]的方法,将10 mg CoTPP配合物催化剂和n(n=1,5,10,15,20)倍质量的GO加入到100 mL的圆底烧瓶中,并加入30 mL乙醇搅拌均匀,随后加入5%(质量分数)Nafion乙醇溶液作为交联剂,3 h后过滤固体并用盐酸洗涤除去未反应物质,剩余固体产物置于真空干燥箱中烘干,即得5,10,15,20-四(苯基)卟啉钴/GO(CoTPP/GO).
1.2.5 CoTPP(SPh2)/GO的制备 采用与CoTPP/GO相同的合成方法和复合比例制备CoTPP(SPh2)/GO.
2 结果与讨论
2.1 电催化析氢复合材料的制备与表征
图1(A)为TPP,CoTPP和CoTPP(SPh2)的UV-Vis谱图.可见,TPP在420 nm处有卟啉化合物典型的Soret带和500~700 nm范围内4个微弱吸收峰的Q带,与中心金属Co配位后,CoTPP的Q带个数减少,CoTPP(SPh2)的Soret带吸收峰位置发生红移,表明SPh2已经连接在CoTPP轴向位置上.图1(B)为卟啉石墨烯基复合材料的UV-Vis DRS光谱,发现CoTPP/GO复合材料比CoTPP卟啉催化剂的Soret带和Q带明显展宽,表明金属卟啉与石墨烯间有强的π-π堆积作用[31].CoTPP(SPh2)配合物的高分辨质谱(图S1,见本文支持信息)和元素分析(图S2,见本文支持信息)进一步证明所得产物为目标化合物.

Fig.1 UV⁃Vis spectra of TPP,CoTPP,CoTPP(SPh2)(A)and UV⁃Vis DRS spectra of the GO,CoTPP/GO,CoTPP(SPh2)/GO(B)
首先对CoTPP与GO的质量比进行了优化.由图2的线性扫描伏安(LSV)曲线可见,不同配比的CoTPP/GO(1∶1,1∶5,1∶10,1∶15,1∶20)复合材料对应的析氢起始电位分别为−528,−410,−370,−337和−360 mV.结果显示,复合材料CoTPP/GO最佳比例为1∶15(图S3,见本文支持信息),以下复合材料的表征和电化学测试,未特殊说明均采用CoTPP∶GO=1∶15质量比的复合材料.
图3(A)和(B)分 别 给 出 了CoTPP/GO和CoTPP(SPh2)/GO中Co2p的XPS谱 图.与CoTPP(图S4,见本文支持信息)相比,复合材料CoTPP/GO的Co2p1/2轨道结合能由794.6 eV增至794.9 eV,而Co2p3/2对应的轨道结合能由779.2 eV提高到779.8 eV,表明CoTPP与GO发生了相互作用.同样与CoTPP(SPh2)相比,CoTPP(SPh2)/GO的Co元素结合能也发生了类似的相互作用,Co2p1/2轨道的结合能由794.8 eV增至795.2 eV,Co2p3/2轨道的结合能由779.3 eV提高到780.2 eV.

Fig.2 LSV curves of different mass ratio of CoTPP and GO
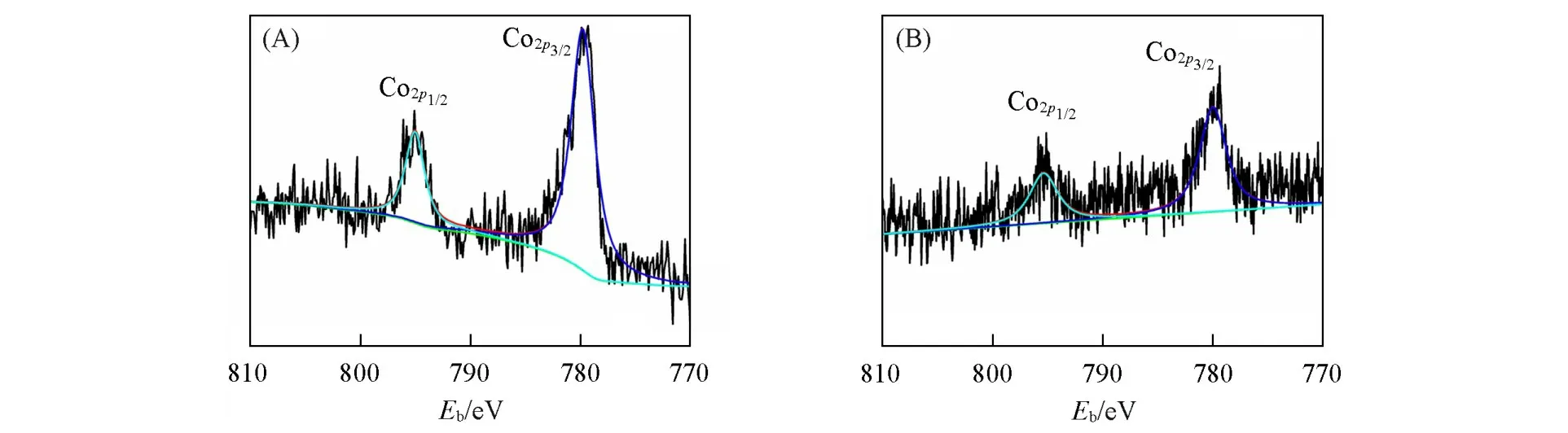
Fig.3 XPS spectra of Co2p of CoTPP/GO(A)and CoTPP(SPh2)/GO(B)
图4 为材料的SEM照片.可见,未经改性的GO[图4(A1)和(A2)]是高度有序的致密层状堆积结构,这种有序堆积的结构不利于电极表面电子在复合材料内部的迁离转移,材料内部阻抗大,导电性能低.经过自组装修饰的复合材料CoTPP/GO表面无序程度明显增加[图4(B1)和(B2)],表面结构缺陷性更大[32],可以有效抑制复合材料电子空穴的复合,使电子在材料内部持续地迁离转移传递到材料的催化活性位点.而加入轴向配体的CoTPP(SPh2)/GO复合材料表面无序度进一步增加[图4(C1)和(C2)],更有利于提高电催化效率.

Fig.4 SEM images of GO(A1,A2),CoTPP/GO(B1,B2)and CoTPP(SPh2)/GO(C1,C2)with different magnifications
图5 为材料的Raman光谱图.GO在1346和1589 cm−1处的吸收谱带是石墨碳材料典型的D峰与G峰.D峰是由碳材料本身结构的缺陷、边缘不规整及结构的无序性导致产生的,而G峰主要是碳材料中含有sp2有序碳的E2g模型结构导致形成的.与GO相比,CoTPP/GO和CoTPP(SPh2)/GO复合材料的D峰与G峰位置基本不变.GO的D峰与G峰分别反映其结构缺陷与sp2碳原子的面内振动[33].CoTPP/GO的D峰与G峰强度比值(ID/IG)相比于GO的ID/IG比值由0.82增至0.86,表明GO表面的结构缺陷增多,这是由于金属卟啉与石墨烯边缘带隙作用所致,表明CoTPP/GO复合材料的表面无序程度比纯GO有所增加.同样,修饰轴向配体的CoTPP(SPh2)/GO复合材料的ID/IG值增至0.89,更加有利于电子在复合材料内部迁离转移到材料表面活性位点还原氢质子,因此这种复合材料有利于提高电催化析氢性能.

Fig.5 Raman spectra of GO,CoTPP/GO and CoTPP(SPh2)/GO
2.2 电化学析氢性能
未经改性的GO析氢起始过电位为−761 mV,电流密度1 mA/cm2对应的过电位为−764 mV[图6(A)],同时析氢曲线对应的动力学传质过程十分缓慢,然而具有钴金属中心的CoTPP金属化合物电催化剂表现出良好的动力学传质性质,同时析氢起始电位降低为−557 mV,电流密度1 mA/cm2对应的过电位达到−561 mV,而自组装改性的CoTPP/GO复合材料的析氢起始电位明显下降至−337 mV,电流密度1 mA/cm2对应的过电位降低至−343 mV,我们认为GO与CoTPP间的堆积作用增加了材料间的电荷转移,提高了复合材料的导电性能.加入二苯硫醚轴向配体后,CoTPP(SPh2)/GO材料的析氢起始电位进一步下降至−235 mV,电流密度1 mA/cm2对应的过电位达到−239 mV,表明轴向二苯硫醚配体的加入可以进一步加快电子与质子源的结合速率,提高氢质子还原过程的效率.这些结果证实材料自组装和轴向配体作用可以显著提高复合材料的电催化析氢性能.

Fig.6 Polarization curves(A)and Tafel plots(B)of HER process for GO,CoTPP,CoTPP(SPh2),CoTPP/GO,CoTPP(SPh2)/GO in 0.5 mol/L H2SO4 aqueous solution
超电势(η)与电流密度(i)存在如下关系:η=a+blg|i|(其中,a和b为塔菲尔常数).可见,超电势与电流密度对应的塔菲尔曲线斜率越小,Volmer-Heyrovsky[34]动力学传质过程越快.图6(B)为GO,CoTPP,CoTPP(SPh2),CoTPP/GO和CoTPP(SPh2)/GO的塔菲尔曲线,未经改性的GO塔菲尔曲线斜率为296 mV/dec,电子传递过程速率缓慢,而卟啉钴CoTPP和CoTPP(SPh2)配合物的塔菲尔曲线斜率则分别为209和197 mV/dec,而经过自组装的CoTPP/GO复合材料塔菲尔曲线斜率明显降低为174 mV/dec,表明CoTPP与GO的π⁃π堆积作用和轴向配体作用可以进一步加快电子交换的速率,加入轴向配体塔菲尔曲线斜率进一步降低为163 mV/dec,电催化产氢性能更好.
交流阻抗谱图(Electrochemical Impedance Spectroscopy,EIS)由高频率的半圆部分和低频率的直线部分组成.高频率下的半圆部分对应于电子转移限制过程,半圆的直径代表电荷转移阻抗Rct,常用于评价不同材料的导电性能和电子传递效率[35].由图7可见,未经处理的GO的电荷转移阻抗Rct达到1180 Ω,而经过自组装改性的CoTPP/GO材料的Rct下降至734 Ω,这主要是由于CoTPP与GO之间的相互作用可以有效地加快材料体系内的电子转移速率,使其具有更好的电荷转移能力,复合材料CoTPP/GO的电荷转移阻抗明显降低.加入轴向二苯硫醚配体的CoTPP(SPh2)/GO复合材料的Rct进一步降至580 Ω,复合材料的阻抗进一步降低,表明CoTPP(SPh2)/GO复合电催化材料的内部阻抗进一步降低,电子从电极内部穿过复合材料内部的迁离和转移速率更快,电催化产氢性能更加优良.
通过在−0.4~1 V的无析氢析氧的电位区间循环扫描1000次,对比两次线性伏安扫描曲线的析氢起始电位以及动力学传质过程的变化,以此测试这种复合材料的稳定性.经历过1000次循环扫描后,CoTPP/GO(图S5,见本文支持信息)和CoTPP(SPh2)/GO[图8(A)]复合材料的析氢起始电位没有变化,动力学曲线向阴极电位方向稍稍偏移,没有明显的催化剂分解现象,在严苛和大量的电极循环条件下电解催化剂的性能没有明显失活,表明该复合材料催化剂的稳定性良好[36].
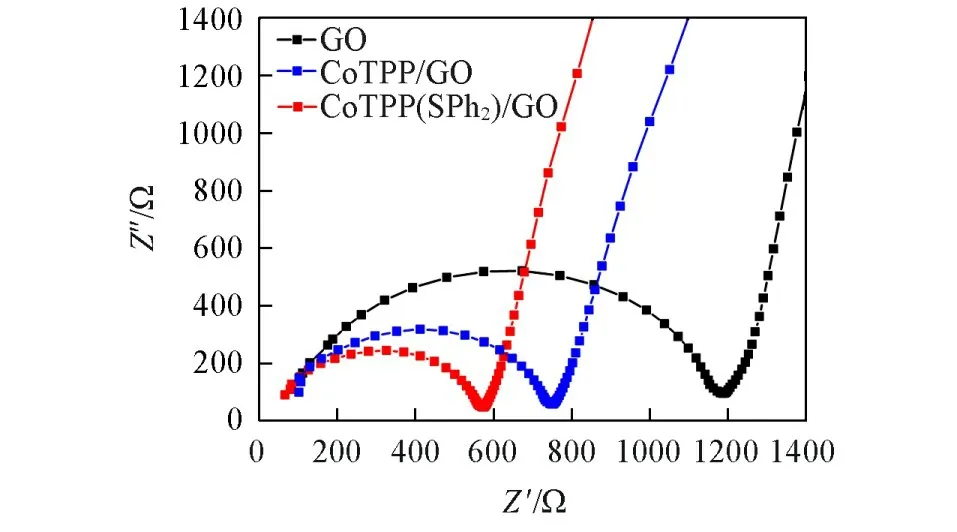
Fig.7 EIS curves of GO,CoTPP/GO and CoTPP(SPh2)/GO

Fig.8 Stability curves(A)and gas chromatogram(B)of hydrogen by CoTPP(SPh2)/GO
法拉第效率(Faraday Efficiency,FE)是用实际生成物消耗量与理论生成物消耗量的比值来表征催化转换效率[37].图8(B)为CoTPP(SPh2)/GO的电催化析氢的气相色谱气体收集图,在0.5 mol/L H2SO4电解槽溶液中在1.45 V电压下恒电位电解1 h(图S6,见本文支持信息),用CH4做内标,气相色谱仪收集电解产生的氢气收集曲线,用实际生成氢气需要的电量和理论上消耗的电量的比值来表明电解制氢转换效率.相比于未经处理的GO电解析氢的法拉第效率23%(图S7,见本文支持信息),经自组装后的复合材料CoTPP/GO电解析氢的法拉第效率达到87%(图S8,见本文支持信息),表明电解水制氢反应过程中转换效率较高.CoTPP(SPh2)/GO法拉第效率进一步提升至94%.这是因为金属卟啉和轴向配体作用可以有效促进反应体系中心金属电荷的转移从而增强复合材料内的电子交换速率.
综上所述,本文利用5,10,15,20-四(苯基)卟啉钴与氧化石墨烯自组装构筑了CoTPP/GO电催化析氢复合材料.自组装后复合材料表面的无序度增加,阻抗也明显降低,表明π⁃π堆积作用有助于提高钴卟啉与氧化石墨烯间的电子传输效率,使电极内部电子迅速传递到修饰电极表面复合材料的催化活性位点,实现氢质子的迅速还原电催化转化,从而提高电催化析氢性能.体系中加入二苯硫醚轴向配体后,材料CoTPP(SPh2)/GO的交流阻抗进一步降低,最终电催化析氢起始电位降低为−235 mV,塔菲尔斜率降低至163 mV/dec,法拉第效率达到94%,揭示了轴向配体可以进一步提升复合材料的电催化转换效率.这些结果表明,通过氧化石墨烯与金属卟啉的非共价键自组装是构筑石墨烯基电催化产氢材料的一条有效途径.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20210549.
猜你喜欢
粉末冶金技术(2021年3期)2021-07-28 06:26:50
农药科学与管理(2019年8期)2019-11-23 08:04:44
中国有色金属学报(2018年2期)2018-03-26 07:58:37
童话世界(2017年29期)2017-12-16 07:59:32
中南大学学报(自然科学版)(2016年2期)2017-01-19 07:37:25
中学生数理化·高二版(2016年6期)2016-05-14 13:19:33
中国资源综合利用(2016年7期)2016-02-03 03:00:13
化学工业与工程(2015年1期)2015-02-10 03:01:33
应用化工(2014年11期)2014-08-16 15:59:13
无机化学学报(2014年12期)2014-02-28 17:34:01
