
溶酶体与神经退行性疾病
2022-02-21 07:29:32曲莉丽仓春蕾
生物学杂志 2022年1期
曲莉丽,仓春蕾
(中国科学技术大学 基础医学院,合肥 230026)
溶酶体是真核细胞内发挥降解功能最主要的细胞器,它含有60多种水解酶,可以降解由内吞、自噬等途径输送到溶酶体的多种生物大分子和损伤的细胞器,并将降解产物运送到细胞内或细胞外重新利用。因此,自从1955年Duve等[1]发现溶酶体以来,在很长一段时间里溶酶体都被看作是细胞内的“回收站”。但近年来的研究发现,除了降解功能外,溶酶体还在细胞的营养感知、信号转导、基因表达调控、细胞死亡、质膜修复、抗原加工和呈递以及细胞器质量控制等过程中发挥重要作用[2]。因此,溶酶体的功能异常对细胞和机体有多方面的影响,也和多种疾病密切相关,如溶酶体贮积症、神经退行性疾病、炎症、自身免疫疾病、肿瘤等[3]。
溶酶体贮积症是一大类由于溶酶体相关基因突变引起的单基因遗传性疾病,目前已知的相关基因有70多个,这些基因编码溶酶体酶、溶酶体膜蛋白或溶酶体功能调控因子,它们的突变会导致溶酶体功能异常,引起降解底物在溶酶体中大量积累,最终导致细胞功能障碍和细胞死亡[4]。根据贮积物和病理特征的不同,溶酶体贮积症可以分为不同的类型。常见的溶酶体贮积症有戈谢氏病(Gaucher disease)、法布里病(Fabry disease)、尼曼匹克病(Niemann-Pick disease)、黏脂质贮积症(mucolipidosis)、神经元蜡样脂褐质沉积症(neuronal ceroid lipofuscinosis)等。由于神经元对溶酶体贮积更为敏感,而且神经元是终末分化的细胞,一旦死亡后无法得到有效的补充,因此大部分溶酶体贮积症都出现神经系统退行性病变的症状,这一现象也将溶酶体与神经退行性疾病联系起来[5]。
神经退行性疾病是一类由神经元结构或功能逐渐丧失甚至死亡而引起神经系统功能障碍的疾病,包括阿尔茨海默病(Alzheimer’s disease,AD)、帕金森病(Parkinson’s disease,PD)、亨廷顿病(Huntington’s disease,HD)、肌萎缩侧索硬化症(amyotrophic lateral sclerosis,ALS)、神经元蜡样脂褐质沉积症(neuronal ceroid lipofuscinosis,NCL)、额颞叶痴呆(frontotemporal lobar dementia,FLTD)等多种类型。除了少数家族性神经退行性疾病有明确的致病基因外,大多数疾病的发病机制尚不清楚,因此缺乏有效的治疗手段[6]。
近年来,随着研究的逐步深入,越来越多的证据表明溶酶体功能异常与神经退行性疾病密切相关。首先,在很多神经退行性疾病的病人中都发现了溶酶体功能的改变。有统计显示在英国儿童发生的神经退行性疾病中,约有45%的病人存在溶酶体功能异常[7]。其次,针对神经退行性疾病的基因组关联分析也发现了多个溶酶体相关基因,进一步确认了溶酶体与神经退行性疾病的关系[8-9]。另外,神经退行性疾病中常出现物质的异常累积,也提示作为细胞“回收站”的溶酶体的降解功能可能出现了异常。最后,溶酶体还被发现是衰老的重要调控位点,而衰老是多种神经退行性疾病的关键诱发因素[10]。因此,研究溶酶体功能异常在神经退行性疾病中的作用,有助于进一步解析神经退行性疾病的发病机制,也有望为疾病的治疗和药物开发提供新的靶点。
溶酶体功能异常可能是由多方面因素导致的,如溶酶体酶的表达和活性改变、溶酶体pH稳态失衡、离子通道或其他溶酶体膜蛋白功能异常、溶酶体定位异常等,这些因素与神经退行性疾病的关系也得到了广泛研究。
1 溶酶体酶
溶酶体的降解功能是由60多种溶酶体酶完成的,这些酶包括糖苷酶、蛋白酶、核酸酶、酯酶等不同类型,分别负责特定底物的分解代谢。除了降解功能外,溶酶体酶在蛋白质加工、溶酶体稳态维持、细胞凋亡以及免疫应答等生理过程中都发挥至关重要的作用。溶酶体酶的功能缺陷可导致相应的底物无法降解从而在溶酶体中大量积累,引起细胞代谢和胞内信号通路紊乱,进而导致溶酶体贮积症[4]。这些溶酶体贮积症中有很多种都表现出神经病理学症状[5]。
事实上,在常见的神经退行性疾病中经常发现溶酶体酶活性的改变。在众多的溶酶体酶中,组织蛋白酶cathepsin是最受关注的类型,它与衰老和神经退行性疾病关系也被逐步揭示出来。衰老的一个重要特征是蛋白质的稳态失衡,而负责降解蛋白质和多肽的cathepsin的功能异常则是引起蛋白质稳态失衡和衰老的重要原因[11]。cathepsin有多种不同的类型,可以分为3个亚群:丝氨酸蛋白酶,包括cathepsin A和G;天冬氨酸蛋白酶,包括cathepsinD和E;半胱氨酸蛋白酶,包括cathepsin B、C、F、H、K、L、O、S、V、X和W。在这些酶中有很多种都与衰老和神经退行性疾病相关。
研究发现在衰老大鼠的脑组织中,cathepsin L的蛋白水平未改变,但其活性却显著降低[12]。另一项研究则发现cathepsin B也存在类似的情况[13]。cathepsin B和L对小鼠出生后中枢神经系统的发育至关重要,这两种酶缺失的小鼠出现脑内特定神经元的大量凋亡并表现出一定程度的脑萎缩[14]。与B和L不同的是,cathepsin D和E在衰老的大鼠和人的多个脑区中均出现表达水平和活性的升高[12,15],这可能还与衰老神经细胞中脂褐质的积累有关[16]。
与溶酶体酶最直接相关的神经退行性疾病是神经元蜡样脂褐质沉积症(NCL)。NCL是一类早发型神经退行性疾病,大部分患者在婴幼儿期发病,症状包括痴呆、失明、脑萎缩、癫痫、早亡等,细胞学特征为自发荧光脂褐质和ATP合酶亚基C在溶酶体的积累,因此也是溶酶体贮积症的一种[17]。NCL疾病是由十几种CLN基因的突变导致的,其中CLN1、CLN2、CLN10和CLN13基因均编码溶酶体酶。CLN1基因编码溶酶体酶PPT1(palmitoyl protein thioesterase-1),这种酶负责从蛋白质的半胱氨酸位点上切割长链脂肪酸残基。CLN2基因编码溶酶体酶TPP1(tripeptidyl peptidase 1),它本身是无活性的酶原,在溶酶体中活化,负责将蛋白和多肽切割为三肽。CLN10和CLN13基因分别编码cathepsin D和F,因此这两种蛋白也被称为CLN10和CLN13[18]。
多种常见神经退行性疾病的标志性蛋白是溶酶体酶的降解底物或产物,如β-淀粉样蛋白(β-amyloid, Aβ)、α-突触核蛋白(α-synuclein)、亨廷顿蛋白(huntingtin)等,而溶酶体酶的异常,尤其是组织蛋白酶的表达和活性改变也常见于这些疾病中。
阿尔茨海默病(AD)是最常见神经退行性疾病,表现为进行性的认知功能障碍和行为损害,包括记忆障碍、语言和运动能力减退、人格和行为异常等多方面症状。这些症状主要是由于大量神经元死亡及神经突触的丢失引起的,但其机制尚不完全清楚。脑中Aβ积累产生的淀粉样斑块和Tau蛋白过度磷酸化形成的神经纤维缠结是AD的典型病理特征,也被认为是引起神经细胞死亡的可能原因[19]。Aβ是由β-secretase和γ-secretase这两种酶对APP(amyloid precursor protein)蛋白进行切割产生的,而溶酶体被认为是切割的主要位点之一[20]。β-secretase的活性在pH 4.5左右,是溶酶体的pH值[21]。γ-secretase需要和AD相关蛋白Presenilin 1(PS1)及其他几个蛋白组成复合体发挥酶切活性[22]。PS1编码基因的突变是导致家族性AD的最主要原因,而且PS1突变也会影响自噬流以及溶酶体酶的活性[23]。
一些溶酶体酶可能与Aβ的生成及降解相关,如cathepsin B和D,但相关的研究存在较大争议。早期研究发现AD病人中cathepsin D的表达和活性均有所上升,并且存在于淀粉样斑块中[24-25],因此认为cathepsin D和AD相关。研究表明cathepsin D基因的核苷酸多态性与AD的发病率相关[26],但随后一些研究却不支持这一观点[27-28]。cathepsin D也被认为具有类似于β和γ-secretase类似的切割APP的活性[29],但也有研究在cathepsin D缺陷的海马神经元中并未发现APP切割的异常[30]。除了Aβ外,Tau蛋白也可能与cathepsin D相关,有研究发现cathepsin D的抑制剂可以抑制AD中Tau的过度磷酸化现象[31]。cathepsin B最早被认为可以有效降解Aβ[32-33],但也有研究发现抑制cathepsin B反而可以降低Aβ的积累并改善AD模型小鼠的记忆[34]。总之,AD和cathepsin的关系还有待进一步研究。
帕金森病(PD)是仅次于AD第二高发的神经退行性疾病,主要症状为震颤麻痹。PD的发病原因为中脑黑质多巴胺能神经元的死亡,但除了少部分家族性PD是由几种相关基因突变引起的之外,大部分散发型PD的发病机制目前尚不清楚[35]。α-突触核蛋白的积累和路易小体(Lewy bodies)的出现是PD的典型病理特征,提示蛋白质稳态出现异常。cathepsin D是降解α-突触核蛋白的最主要的溶酶体酶[36]。cathepsin D缺陷小鼠的脑中出现α-突触核蛋白的过度积累,而过表达cathepsin D的细胞则可以促进α-突触核蛋白的降解并减轻其带来的毒性[37-38]。除了cathepsin D外,也有研究提示cathepsin B和L和α-突触核蛋白的沉积或降解相关[39]。
亨廷顿病(HD)是一种huntingtin基因突变导致的遗传性神经退行性疾病,症状包括运动协调和认知受损、肌张力障碍以及舞蹈样动作等,因此也常被称为亨廷顿舞蹈症。Huntingtin基因编码的Huntingtin蛋白是cathepsin D、B和L的底物,这些酶参与Huntingtin蛋白的加工[40]。正常的亨廷顿蛋白可以被cathepsin D降解,但突变的亨廷顿蛋白则对cathepsin D有更高的抗性[41]。在亨廷顿病人脑组织中发现cathepsin D活性的增加,这一现象在体外细胞实验中也得到验证[41-42]。这种活性增强可能并不是促进疾病发生的因素,因为有研究发现过表达cathepsin B和D在HEK293细胞及原代培养神经元中均表现出神经保护的作用[43]。
肌萎缩侧索硬化(amyotrophic lateral sclerosis,ALS)又叫运动神经元病或“渐冻病”,主要症状为进行性运动神经元丢失及肌无力和肌萎缩,导致病人在发病后3~5年内死亡。大部分ALS病人的发病机制目前尚不清楚。在ALS模型小鼠的脊髓中,cathepsins B、L、D和S的表达均有明显上升[44-45],而在ALS病人中则仅有cathepsins B和D出现表达上调[46]。
除了组织蛋白酶外,其他溶酶体酶的异常也在神经退行性疾病中被发现。如在散发型帕金森病人脑组织中β-葡糖脑苷脂酶(β-glucocerebrosidase)的表达水平和酶活性的降低,并与溶酶体分子伴侣介导自噬的减少、α-突触核蛋白的异常积累以及神经酰胺的减少直接相关[47]。β-葡糖脑苷脂酶编码基因GBA1的突变本身就是帕金森病和路易体痴呆病的危险因素,也是最常见的一种溶酶体贮积症——戈谢氏病(Gaucher’s disease)的主要诱发因素[48]。另一项研究在帕金森病人脑脊液中也发现β-葡糖脑苷脂酶活性的降低,同时发现另一个溶酶体酶β-己糖胺酶(β-Hexosaminidase)的活性反而升高了。而在亨廷顿舞蹈病患者的脑脊液中则发现酸性磷酸酶(acid phosphatase)的活性增高,且与年龄正相关[49]。
这些研究表明神经退行性疾病与溶酶体酶功能异常相关(表1),因此溶酶体酶逐渐成为这些疾病药物开发的新靶点。一些针对溶酶体酶的替代疗法已经得到临床应用,大量的小分子药物也在开发中,有望为神经退行性疾病的治疗和药物开发带来新的曙光[50-51]。

表1 神经退行性疾病与相关的溶酶体酶
2 溶酶体pH和V-ATPase
大多数溶酶体酶是酸性水解酶,因此溶酶体腔内需要维持pH值为4.5~5.0的酸性环境[52]。这种酸性环境还是溶酶体钙离子储存、囊泡运输、营养感知、信号转导以及维持溶酶体完整性的基本条件,对溶酶体的功能至关重要。近年来大量研究发现溶酶体酸化改变是衰老的重要调控因子[53]。如多项酵母中的研究发现衰老过程常常伴随着溶酶体pH值的升高,加强溶酶体酸化是酵母复制寿命延长的关键因素之一[54-55]。而在人类中,溶酶体酸化缺陷已被发现与多种神经退行性疾病密切相关[56],恢复溶酶体pH被认为是治疗这些疾病的重要策略。如Coffey等[57]发现在PS1突变引起的家族性AD患者的皮肤成纤维细胞中,溶酶体 pH 值有小幅度但显著的增加,并伴有溶酶体内成熟组织蛋白酶D及其活性位点水平的降低以及自噬底物的大量积累。一些与神经退行性疾病的致病基因编码的蛋白也被证明与溶酶体pH调控相关,如帕金森病(PD)相关蛋白LRRK2(leucine rich repeat kinase 2)、TMEM175(transmembrane protein 175)、ATP13A2(ATPase cation transporting 13A2)[56,58-59],额颞叶型痴呆(FTD)相关蛋白TMEM106B(transmembrane protein 106B)[60]、肌萎缩性脊髓侧索硬化症(ALS)相关蛋白UBQLN2(ubiquilin 2)等[61],这或许是这些疾病中溶酶体功能异常的潜在机制。
溶酶体酸化主要是靠溶酶体膜上的质子泵V-ATPase实现的,它通过水解ATP产生能量将胞质侧的H+泵入溶酶体腔内[52]。V-ATPase是一种复杂的多亚基蛋白复合物,由细胞质侧的V1区和溶酶体膜锚定的V0区组成。哺乳动物V-ATPase的V1结构域由A~H 8种类型的亚基组成,V0结构域则包括a、c、c′、c″、d、e等6种不同的亚基[62](图1)。在V1结构域中,3个V1A与3个V1B亚基组成异六聚体介导 ATP的结合和水解,并驱动由V1D与V1F形成的中央转子亚复合体的旋转导致H+逆浓度梯度转运通过V0区域进入溶酶体腔内[62]。
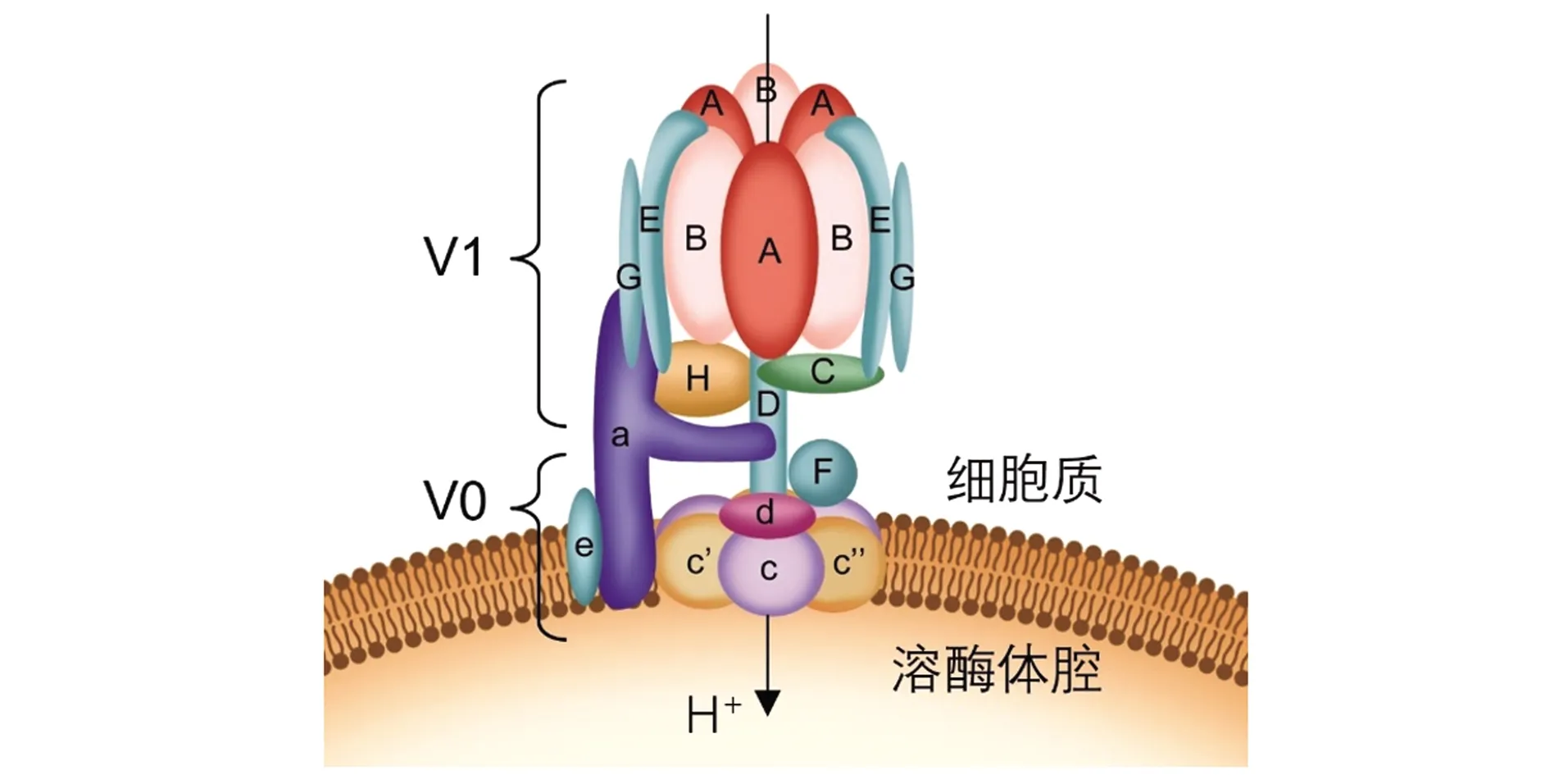
图1 溶酶体质子泵:V-ATPase
研究表明V-ATPase与衰老及多种神经退行性疾病相关,它组成亚基的遗传缺陷可以直接导致多种神经系统功能异常。V1B2在大脑中高表达,它的突变会损害溶酶体酸化并导致感觉神经性耳聋[63]。V1B1及V0a4亚基功能缺失性突变均会造成婴幼儿生长迟缓、佝偻病以及神经性听力丧失[64]。V-ATPase还参与视觉功能的正常发育,V0a3亚基突变会导致人类严重的常染色体隐性骨硬化症,V0a3缺乏的小鼠出现视神经管受压造成视神经损伤,在出生2~3周之后即发生视网膜退化[65]。先天性糖基化病及常染色体隐性皮肤松弛症与V0a2亚基功能丧失相关,这些病人往往容易出现大脑发育异常、智力低下、痴呆、癫痫等症状[66]。
除了V-ATPase编码基因本身的突变外,V-ATPase亚基蛋白的表达水平和稳定性改变也是神经退行性疾病中的常见现象。如AD相关蛋白PS1除了在淀粉样前体蛋白APP生成淀粉样蛋白Aβ中起重要作用外,还是V-ATPase V0a1 亚基的特定配体,对V-ATPase 靶向溶酶体、溶酶体酸化和自噬过程中的蛋白水解至关重要[23]。PS1 突变的家族性AD患者细胞和PS1敲除的小鼠细胞均表现出 V0a1 成熟缺陷,同时散发型AD病人海马神经元中V0a1的mRNA表达也有所降低,引起V-ATPase活性降低、溶酶体pH值升高、水解能力降低,进而引起的自噬功能障碍而无法降解大量聚集的Aβ[23,57,67]。另外,在AD小鼠模型中发现了V1E1、V1A亚基表达水平的降低[68-69]。PD相关蛋白LRRK2会结合V0a1亚基,增强其稳定性和表达水平,而LRRK2致病性的突变R1441C会打破它们之间的结合,降低V0a1的表达,引起溶酶体pH值的升高[59]。FTD/ALS相关蛋白TMEM106B和UBQLN2也是通过影响V0结构域的表达进而造成溶酶体的严重酸化缺陷[60-61]。
3 溶酶体膜蛋白
溶酶体膜上除了上述的质子泵外,还有很多其他类型的蛋白质,包括离子通道、转运体、结构性蛋白(LAMP、LIMP等)、运输复合体原件、膜嵌合的酶以及一些功能尚不清楚的蛋白质。这些膜蛋白分别参与不同的溶酶体功能,它们的功能异常会导致溶酶体功能障碍,进而引起多种类型的疾病,同时也与多种神经退行性疾病相关(图2)。

图2 溶酶体膜蛋白与神经退行性疾病
3.1 溶酶体离子通道
溶酶体的氢离子转运会受到其他离子分布的影响,同时溶酶体还是细胞内钙、铁、锌等离子的重要储存位点,因此溶酶体的离子平衡和转运对溶酶体功能至关重要[70]。溶酶体膜上存在多种类型的离子通道,这些离子通道是调控溶酶体离子平衡的关键蛋白,其中部分离子通道和神经退行性疾病密切相关,包括TRPML1、TMEM175、TPC2和CLC通道等。
TRPML1是溶酶体膜上的一种非选择性阳离子通道,参与调控溶酶体的钙稳态、金属离子储存、溶酶体运输、自噬等[71]。TRPML1的基因突变会导致四型黏脂沉积症(Mucolipidosis type IV,ML-IV),这是一种儿童期发病的神经退行性疾病,主要症状为智力低下和视网膜退行性病变[72]。在TRPML1功能缺陷的细胞中存在溶酶体胀大、脂类物质积累以及溶酶体定位的异常,表明溶酶体降解功能和运输机制受损[73]。除了ML-IV外,TRPML1也和其他几种神经退行性疾病相关。在AD的模型中,TRPML1的表达水平降低,进而影响溶酶体内的钙离子释放及细胞的自噬水平,而过表达或激活TRPML1则会促进β-淀粉样蛋白的清除,减轻神经元的损伤[74-75]。同样,在ALS的小鼠模型中TRPML1的表达也减少了,药物激活TRPML1可以减轻运动神经元的死亡[76]。此外,在尼曼匹克病中,溶酶体中积累的鞘磷脂也会抑制TRPML1的活性,提示TRPML1可能参与到这一疾病中[77]。
TMEM175是新发现的一种溶酶体钾离子通道。钾离子是溶酶体外最主要的阳离子,因此TMEM175对溶酶体功能有多方面的影响,如溶酶体膜电位、pH以及溶酶体和自噬体的融合[58]。在TMEM175的功能被鉴定之前,它和帕金森病之间的联系已经被发现,研究采用全基因组关联分析发现TMEM175基因的单核苷酸多态性突变与帕金森病的发病率及发病年龄相关[78-79],但其机制并不清楚。随后,采用帕金森病的细胞模型,研究发现TMEM175的功能缺失会导致溶酶体和线粒体功能缺陷,促进α-突触核蛋白的积累并增强神经元对α-突触核蛋白的敏感性[80]。最近的一项研究发现TMEM175可以和AKT组成营养因子敏感的通道复合体,其与帕金森病相关的突变则会改变这一通道复合体的活性,其活性降低或功能缺陷也在细胞和小鼠模型中加剧了帕金森病的病理特征[81]。这些研究提示TMEM175有望成为帕金森病治疗和药物开发的一个新靶点。
TPC是溶酶体膜上的钠离子通道,可以和mTOR组成ATP敏感的通道复合体,参与细胞的能量和营养感知以及溶酶体pH的调控[82-83]。也有研究发现TPC通道和NAADP诱发的溶酶体钙离子释放有关[84]。研究发现TPC的一个亚型TPC2可能和帕金森病相关。在携带LRRK2的致病性G2019S突变的细胞中存在溶酶体胀大和聚集现象,而敲低或抑制TPC2可以逆转这种现象,提示TPC2可能是LRRK2的下游效应蛋白,同时也可能成为帕金森病的潜在治疗靶点[85]。
除了这些溶酶体阳离子通道外,一些溶酶体阴离子通道也和神经退行性疾病相关,其中包括多个CLC家族蛋白。CLC家族包含多个成员,其中一部分是氯离子通道,另一部分是则是氯离子/氢离子交换器,而且多个CLC成员是定位在内体/溶酶体膜上的,包括CLC-3、CLC-4、CLC-5、CLC-6和CLC-7[86]。氯离子被认为是平衡溶酶体氢离子转运引起的电荷积累的反离子,因而这些内体溶酶体CLC成员对溶酶体pH的稳定至关重要。CLC-3、CLC-6和CLC-7的功能缺失都会引起神经退行性病变。CLC-3的敲除小鼠在出生后很快就表现出严重的脑和视网膜的退变,在3个月后它们的海马脑区甚至完全丢失[87]。CLC-6敲除小鼠的表型不如CLC-3那么明显,它们在较大鼠龄时才会出现较弱的脑退变及轻微认知障碍[88]。CLC-6的一个功能增强型突变导致了儿童期早发的神经退行性病变[89]。CLC-7是主要定位于溶酶体的亚型,它的膜定位和正常功能需要辅助亚基Ostm1的帮助[90]。CLC-7及Ostm1的功能缺失都可以导致骨硬化症(osteopetrosis),并伴随有脑和视网膜的退行性病变[88,91]。出乎意料的是,CLC-7/Ostm1敲除小鼠的溶酶体,pH并未受到影响,它们的缺陷引起神经退行性病变的机制还有待进一步研究。
3.2 转运体
溶酶体是代谢性细胞器,需要频繁地进行代谢底物和降解产物跨溶酶体膜的运输,因此溶酶体膜上存在多种不同类型的转运体蛋白。这些转运体蛋白的功能异常常常和神经退行性疾病联系起来。ATP13A2(又名PARK9)是溶酶体膜上的一种ATP酶型的转运体蛋白,它通过水解ATP获能跨膜转运重金属离子和多胺类物质[92],其功能异常会影响溶酶体pH、降解功能和自噬流,引起α-突触核蛋白积累和神经毒性[93]。ATP13A2的突变会导致早发型帕金森病和Kufor-Rakeb综合征,这是一种伴有痴呆的帕金森样疾病[94-95]。研究发现ATP13A2的突变和神经元蜡样脂褐质沉积症相关,因此根据这一疾病相关蛋白的命名规则被命名为CLN12[96]。此外,在少数ALS病人中也发现了ATP13A2的突变,进一步说明这一转运体蛋白与神经退行性疾病的高度相关性及其分子机制的复杂性。
NPC1是溶酶体膜上的脂质转运体,NPC2则是溶酶体腔内的脂质结合蛋白,它们协同作用将脂质从溶酶体腔内转运到胞浆或其他细胞器,而它们的突变则会导致溶酶体内胆固醇和鞘脂的积累,进而引起C型尼曼匹克病,这一疾病会引起系统性神经退行性病变[97]。
3.3 结构性蛋白LAMP和LIMP
在所有溶酶体膜蛋白中,LAMP(lysosome-associated membrane protein)是溶酶体膜上含量最多的类型,其中LAMP-1和LAMP-2两种蛋白已占溶酶体膜蛋白总量一半左右[98]。LMAP-1和LAMP-2的功能有一定的互补性,双重缺陷的动物有胚胎致死的表型,而单一蛋白缺陷的表型则弱得多[99]。LAMP-1缺陷的小鼠脑中仅存在轻微的星形胶质细胞增生,并没有发现有明显的脑功能异常,且溶酶体功能也未见显著变化[100]。LAMP-2缺陷小鼠则有更明显的表型,包括自噬囊泡的积累和心肌收缩力减弱,并且有一半左右小鼠在40 d内死亡[101]。LAMP-2缺陷在人类中会导致Danon病,主要表现为心肌病和骨骼肌病,同时伴有神经退行性病变引起的智力障碍和视网膜损伤等[102]。Danon病人的细胞中存在糖原在溶酶体中聚集和自噬囊泡聚集的现象。相比LAMP-1和-2,LAMP-3的表达量较少,但有多个基因组学研究提示它和帕金森病有关[103]。
另一类溶酶体膜结构性蛋白是LIMP(lysosomal integral membrane protein)。LIMP-2蛋白与溶酶体脂质代谢相关,它参与磷脂和胆固醇的转运[104-105],同时它也是β-葡糖脑苷脂酶的受体,协助将β-葡糖脑苷脂酶递送到溶酶体内,因此和戈谢氏病及帕金森病相关[106]。LIMP-2缺陷的小鼠表现出多方面的神经系统异常[107]。
3.4 溶酶体运输复合体原件
VPS35是一种定位于溶酶体/内体膜上的蛋白,它是逆向运输复合体的关键元件,这一复合体介导囊泡系统向高尔基体的逆向运输[108]。VPS35的突变在奥地利的家族性帕金森病人中被发现,从而揭开了它与帕金森病的关系[109]。随后的研究发现VPS35的缺陷会影响溶酶体对α-突触核蛋白的降解并导致线粒体碎片化,进而引起多巴胺能神经元死亡和帕金森病的发生[110-111]。此外,也有研究提示VPS35和阿尔茨海默病相关:在AD病人的海马中,VPS35的表达降低,而VPS35的杂合缺陷小鼠也表现出多种AD的表型,包括Aβ和淀粉样斑块积累、认知记忆障碍、LTP缺陷和突触传递受损等[112];在AD小鼠模型中,药物干预以提高VPS35的表达水平对AD症状也有很好的缓解作用,进一步表明VPS35是神经退行性疾病药物开发的重要靶点。
3.5 溶酶体膜嵌合型酶
除了腔内的可溶性酶外,溶酶体膜上也存在多种膜嵌合的酶。HGSNAT(乙酰肝素-α-氨基葡萄糖苷-N-乙酰基转移酶)就是溶酶体膜嵌合型的酶,它可以催化将乙酰辅酶A的乙酰基转移到肝素-α-氨基葡萄糖苷上,是黏多糖等聚糖降解中的一种重要溶酶体酶。HGSNAT突变导致IIIC型黏多糖病,主要特征为中枢神经系统的退行性病变,症状包括痴呆、视力丧失、耳聋、癫痫、暴躁、失眠等[113]。
3.6 功能未知的溶酶体膜蛋白
溶酶体膜上还存在很多功能尚不清楚的蛋白,其中一些蛋白的编码基因突变也会导致神经退行性疾病的发生,如神经元蜡样脂褐质沉积症的两个致病基因CLN3和CLN7[114-115]。CLN3的氨基酸序列和其他功能已知的蛋白并没有明显的相似性,而CLN7则与MFS转运体超家族存在序列相似性,因此也被命名为MFSD8,它可能是一个转运体蛋白,但其转运底物尚不清楚。这两个基因突变导致的NCL疾病目前还没有很好的治疗手段,因此解析它们的蛋白功能及引起疾病的机制有望为临床治疗和药物开发指明方向。
4 溶酶体的细胞内定位
细胞中通常都包含多个溶酶体。在不同类型的细胞中,溶酶体的数量、大小和分布存在很大差异,而且这些溶酶体在细胞内的分布并非是一成不变的,而是处于动态变化中,且受细胞状态和多种信号的调控[116]。溶酶体可以在马达蛋白的驱动下沿着微管快速移动,其中驱动蛋白(Kinesin)驱动溶酶体的顺行运动,即从核周区域(靠近细胞核)向外周区域(靠近细胞膜)移动,而动力蛋白(Dynein)驱动溶酶体的逆行运动,即从外周区域向核周区域移动。此外,BORC、Arl8、SKIP、Rab7、FYCO1、RILP、ORP1L等蛋白及蛋白复合物和Kinesin/Dynein一起构成溶酶体转运的分子机器[116-117]。溶酶体在细胞内分布与其功能密切相关,处于不同亚细胞区域的溶酶体往往发挥不同的功能。如外周区域分布的溶酶体更多参与细胞膜修复、分泌、营养感知、信号转导等功能,而核周分布的则更多参与自噬[117-118]。此外,溶酶体的快速移动也有助于溶酶体与其他细胞器的互作,如内吞体、自噬体和线粒体等。
神经元是具有特殊形态的细胞,通常包括胞体、树突和轴突这些形态结构上完全不同的部分。溶酶体在神经元不同部位的分布也存在很大差异,一般来说在胞体部分分布最为丰富,而在轴突分布则较少。由于轴突中没有粗面内质网和高尔基体,溶酶体成熟所需的很多物质无法在轴突合成,传统的观点认为轴突中的溶酶体是不成熟的,无法发挥降解功能[119]。轴突以及轴突末梢存在旺盛的内吞和自噬活动,以清除老化和损伤的蛋白和细胞器[120]。自噬体最终要与溶酶体融合以完成降解过程,因此这些轴突内产生的自噬体需要通过逆向运输送到胞体才能与成熟的溶酶体融合[120-121]。但近期也有研究发现胞体部位的成熟溶酶体也会被顺向运输到轴突远端参与局部区域的降解过程[122]。相较于轴突来说,树突中存在更多的溶酶体。溶酶体在树突中的分布受突触信号传递的调控,同时也参与了突触可塑性的调控。如谷氨酸受体的激活会增加树突棘部位的溶酶体数量,进而增强局部的降解能力,并参与对内吞受体的降解[123]。此外,溶酶体的分泌也会影响树突棘形态可塑性的调控[124]。
总之,溶酶体在神经元中的分布和运输对神经元功能至关重要,异常的溶酶体定位和运输与多种神经系统疾病相关。研究人员已经在多种精神类疾病和神经退行性疾病中发现溶酶体定位和运输元件的突变,如溶酶体转运关键蛋白复合物BORC的亚基BORCS7的单核苷酸多态性是导致精神分裂症的一个主要危险因素[125],而dynactin的p150亚基的突变也在部分ALS病人中被发现[126]。在另一种神经退行性疾病SPOAN(spastic paraplegia, optic atrophy and neuropathy)中,kinesin的KLC2亚基的基因上游内含子中存在大片段缺失,进而导致KLC2的过度表达,是导致发病的重要原因[127]。
除了这些溶酶体运输原件外,多种神经退行性疾病相关蛋白也会间接影响溶酶体的运输和定位。亨廷顿病相关蛋白huntingtin是囊泡运输的一个重要调控因子,它可以直接和dynein结合,促进dynein/dynactin介导的囊泡运输[128]。敲低huntingtin会导致溶酶体更多向外周区域分布,而表达huntingtin则会促进溶酶体向核周区域聚集[129]。在亨廷顿病的细胞模型中,突变的huntingtin导致溶酶体向核周区域聚集,并伴随着溶酶体功能的改变、mTORC1的激活以及自噬流的增强[130]。LRRK2(Leucine-rich repeat kinase 2,富亮氨酸重复激酶2)是帕金森病最重要的致病基因,它的突变是导致家族性帕金森病最常见的因素,同时也是散发型帕金森病的危险因子[131]。尽管LRRK2的功能及其致病机制尚未完全清楚,但越来越多的研究发现:LRRK2参与内体和溶酶体的运输及定位的调控;LRRK2蛋白可以定位到溶酶体内体膜上,也可以与早期内体膜蛋白Rab5及晚期内体/溶酶体膜蛋白Rab7相结合,从而影响内体和溶酶体的运输;LRRK2的敲除、过表达以及致病性突变均会改变溶酶体在细胞内的定位[132-133]。
除了这些常见的神经退行性疾病外,一些以神经退行性病变为主要特征的溶酶体贮积症中也存在溶酶体运输和定位的异常,如戈谢氏病、尼曼-匹克病、神经元蜡样脂褐质沉积症、黏脂沉积症等。部分戈谢氏病患者存在严重的神经退行性病理特征,包括神经炎症和神经元丢失等。在这种神经性的戈谢氏病中,研究发现溶酶体的数量和细胞内分布的异常[134]。C型尼曼匹克病(NPC)的主要临床症状包括神经系统的异常,如认知损伤、癫痫、共济失调等。NPC细胞均表现出自噬的异常,这些细胞中的溶酶体也会更集中在核周区域,表明溶酶体分布和运输受到了影响。同时,NPC细胞中溶酶体向轴突的运输也受到了影响,导致轴突自噬受阻和轴突营养不良[135]。另外,神经元蜡样脂褐质沉积症相关蛋白CLN3可以与溶酶体运输动力系统中的多个蛋白相结合,如tubulin、dynactin、dynein、kinesin-2、Rab7、RILP等,进而调控溶酶体在细胞内的定位和运输[136]。而四型黏脂沉积症相关蛋白TRPML1则可以通过释放溶酶体内的钙离子,促进溶酶体向核周区域聚集[73]。
5 总结和展望
人们对溶酶体的认知从最初的“回收站”拓展到信号转导、离子平衡、物质转运和细胞命运决定等多个方面,溶酶体与多种类型疾病的关系也被逐步揭示。目前,靶向调控溶酶体的策略已逐步成为现代药物开发的重要方向,已有多种调控溶酶体功能的小分子化合物被开发出来,包括分子伴侣、V-ATPase抑制剂、酶活性调节剂、离子通道调节剂等,部分化合物展现出很好的药物开发潜力[3]。除了小分子药物外,新型生物技术和生物大分子药物也在溶酶体调控方面得到了应用,例如,酶替代疗法已经用于一些溶酶体贮积症的临床治疗,多项针对溶酶体的基因疗法的开发也已进入临床试验阶段[50]。
随着对溶酶体研究的逐步深入,溶酶体与神经退行性疾病的关系也逐步清晰,越来越多的研究揭示了溶酶体功能异常在多种不同类型神经退行性疾病中的作用。神经退行性疾病的药物开发在过去几十年间经历了大量的失败,而靶向溶酶体治疗神经退行性疾病的新思路有望为这一局面带来改观,大量直接或间接作用于溶酶体相关通路的药物已被用于神经退行性疾病的药物开发[10]。神经细胞不可逆的死亡是神经退行性疾病治疗中最为棘手的问题,而神经干细胞是补充丢失的神经元的重要策略。已有研究发现非特异地激活溶酶体可以使衰老的神经干细胞恢复活性,为中晚期神经退行性疾病的治疗提供了新的思路[137]。
需要注意的是,由于溶酶体分布的广泛性和功能的多样性,干预溶酶体可能对细胞和机体产生多方面的影响,因此靶向溶酶体的药物也可能产生各种副作用。溶酶体还有很强的异质性,同一细胞中不同的溶酶体分别参与不同的功能[138],可能仅仅是部分溶酶体参与了疾病的发生发展中,如何特异性干预特定的溶酶体群体还有待进一步研究。此外,溶酶体位于细胞内,部分溶酶体靶点更是位于溶酶体腔内,如何将药物准确递送到靶点所在的位置也是需要解决的问题。
总之,进一步加强对溶酶体和神经退行性疾病的研究,阐明溶酶体功能异常导致疾病发生的详细机制,鉴定新的溶酶体靶点,设计更有针对性和特异性的药物及干预策略,有望为神经退行性疾病的药物开发带来新的突破。
猜你喜欢
保健医苑(2022年6期)2022-07-08 01:25:28
生物化工(2021年2期)2021-01-19 21:28:13
生物化工(2020年1期)2020-02-17 17:17:58
读与写(2019年35期)2019-11-05 09:40:46
活力(2019年22期)2019-03-16 12:47:04
现代职业教育·高职高专(2018年7期)2018-05-14 16:20:40
老年医学与保健(2017年6期)2017-02-06 05:30:03
中华肩肘外科电子杂志(2017年1期)2017-01-11 03:27:59
中外医疗(2015年5期)2016-01-04 03:57:57
医学研究杂志(2015年5期)2015-06-10 06:43:26
