
高效液相色谱-串联质谱法测定水产品中硝基咪唑类药物残留量
2022-02-21 11:36汤水粉钱卓真位绍红王丽娟
渔业研究 2022年1期
汤水粉,钱卓真,位绍红,王丽娟
(福建省水产研究所,福建 厦门 361013)
硝基咪唑类药物含有5-硝基咪唑环结构,具有抗原虫和抗菌活性,可用于防治滴虫病、球虫病以及六鞭虫病等疾病[1-2],深受养殖户的青睐,在水产养殖中,硝基咪唑主要用于治疗寄生虫的感染,导致该类药物在水产品中残留[3]。由于硝基咪唑及其在生物体内的代谢产物具有潜在的致突变性和致癌性,如果在动物源性食品中残留累积,则会对食品安全构成严重威胁[4-5]。因此,许多国家将硝基咪唑列为违禁药物。我国已将这类药物列入《食用动物禁用的兽药及其他化合物清单(农业农村部公告第250号)》中。欧盟于1993年、1995年和1998年分别禁止洛硝哒唑、地美硝唑和甲硝唑这3种药物用于食用动物;2002年美国食品与药物管理局公布了在进口动物源性食品中禁止使用包括地美硝唑等硝基咪唑类药物在内的共11种药物[6];我国食品中兽药最高残留限量标准规定的只能用于治疗,但不得在动物性食品中检出的药物就包括甲硝唑和地美硝唑[7]。
目前,国内外用于水产品、兽产品等动物源性食品中硝基咪唑类药物及其代谢物的检测方法主要有酶联免疫法[8-9]、高效液相色谱法[10-12]、气相色谱法[13]和液相色谱-质谱联用法等[14-15]。其中高效液相色谱-质谱联用法因灵敏度高、选择性和特异性好、操作简单、能够对低浓度的样品进行很好的定性确认,而被广泛应用于水产品中各类药物残留的检测[16-17]。本研究在已有文献和相关标准的基础上,优化了前处理方法,采用乙酸乙酯提取、正己烷除脂,结合液相色谱-串联质谱高度的选择性和抗干扰能力,建立了水产品中硝基咪唑类药物及其代谢物残留的快速检测方法。该方法的灵敏度高,实验周期短,提高了工作效率。改进后的方法简便、准确,能更好地满足水产品中硝基咪唑及其代谢物检测的要求,适合于大批量样品的检测。
1 材料与方法
1.1 实验材料
实验所用水产品均购自当地农贸市场、超市或养殖户。样品采回后取适量肌肉部分进行均质,所有基质样品于-20℃冷冻保存,待用。
硝基咪唑类药物标准品(德国Dr.Ehrenstorfer),纯度≥98%;甲醇、乙腈、乙酸乙酯、正己烷(美国Tedia),色谱纯;无水硫酸钠(国药集团化学试剂有限公司),分析纯;实验用水为Millipore Academic制备的超纯水。
1.2 仪器与设备
TSQ Quantum Ultra液相色谱-串联四级杆质谱联用仪(美国赛默飞世尔科技公司);MS105电子分析天平、 ME203E/02电子分析天平(梅特勒-托利多国际贸易有限公司);MS3型旋涡混合器(德国IKA公司);离心机(北京时代北利离心机有限公司);旋转蒸发仪(上海申生科技有限公司)。
1.3 样品前处理方法
称取(5.00±0.05) g均质后的样品,置于100 mL的塑料离心管,加入5 mL磷酸二氢钠缓冲液(pH=8.0),涡旋混匀。再加入30 mL乙酸乙酯和3 g无水硫酸钠,涡旋振荡50 s,以3 500 r/min 离心5 min,将上清液转入100 mL鸡心瓶。残渣加入20 mL乙酸乙酯重复提取一次,合并提取液于100 mL鸡心瓶。用旋转蒸发仪于40℃水浴蒸干。准确加入1.0 mL 10%甲醇水溶液,涡旋振荡溶解残留物,再加入1 mL乙腈饱和正己烷涡旋混合30 s,以4 000 r/min离心5 min,取下层清液,经0.22 μm滤膜过滤后,供液相色谱-串联质谱仪测定。
1.4 仪器分析条件
色谱柱:Hypersil Gold C18柱(100 mm × 2.1 mm,3.0 μm,美国 Thermo公司);柱温:30℃;
流动相:A为体积分数0.1%的甲酸水溶液,B为甲醇;流速:0.250 mL/min。
流动相梯度洗脱程序见表1。

表1 流动相梯度洗脱程序Tab.1 Gradient elution program
离子源:电喷雾离子源(ESI),正离子模式;离子传输管温度:350℃;喷雾电压:3.0 kV;汽化温度:200℃;鞘气(氮气)压力:45 arb;辅助气(氮气)压力:10 arb;碰撞气(高纯氩气)压力:1.5 mTorr。10种硝基咪唑类药物的质谱参数见表2。
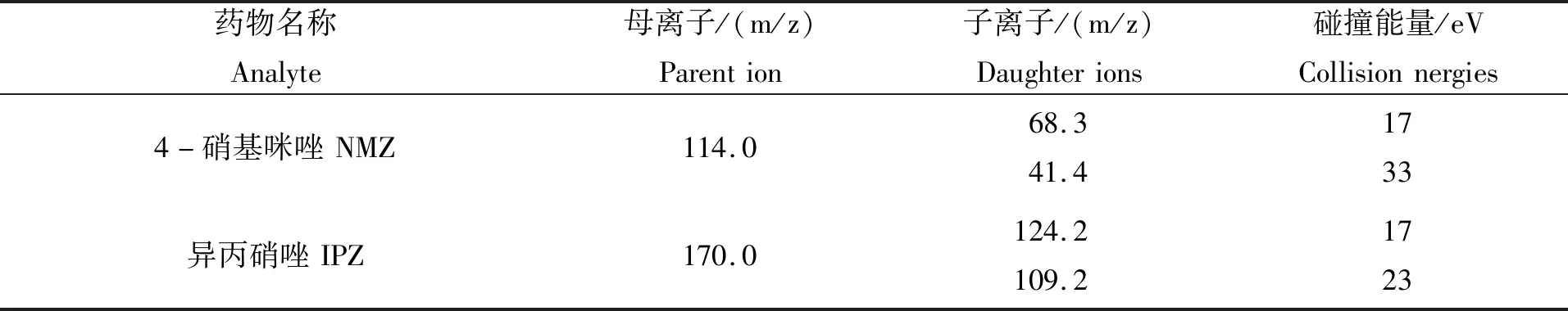
表 2 硝基咪唑类药物及其代谢物的质谱参数Tab.2 MS parameters of nitroimidazoles and their metabolites
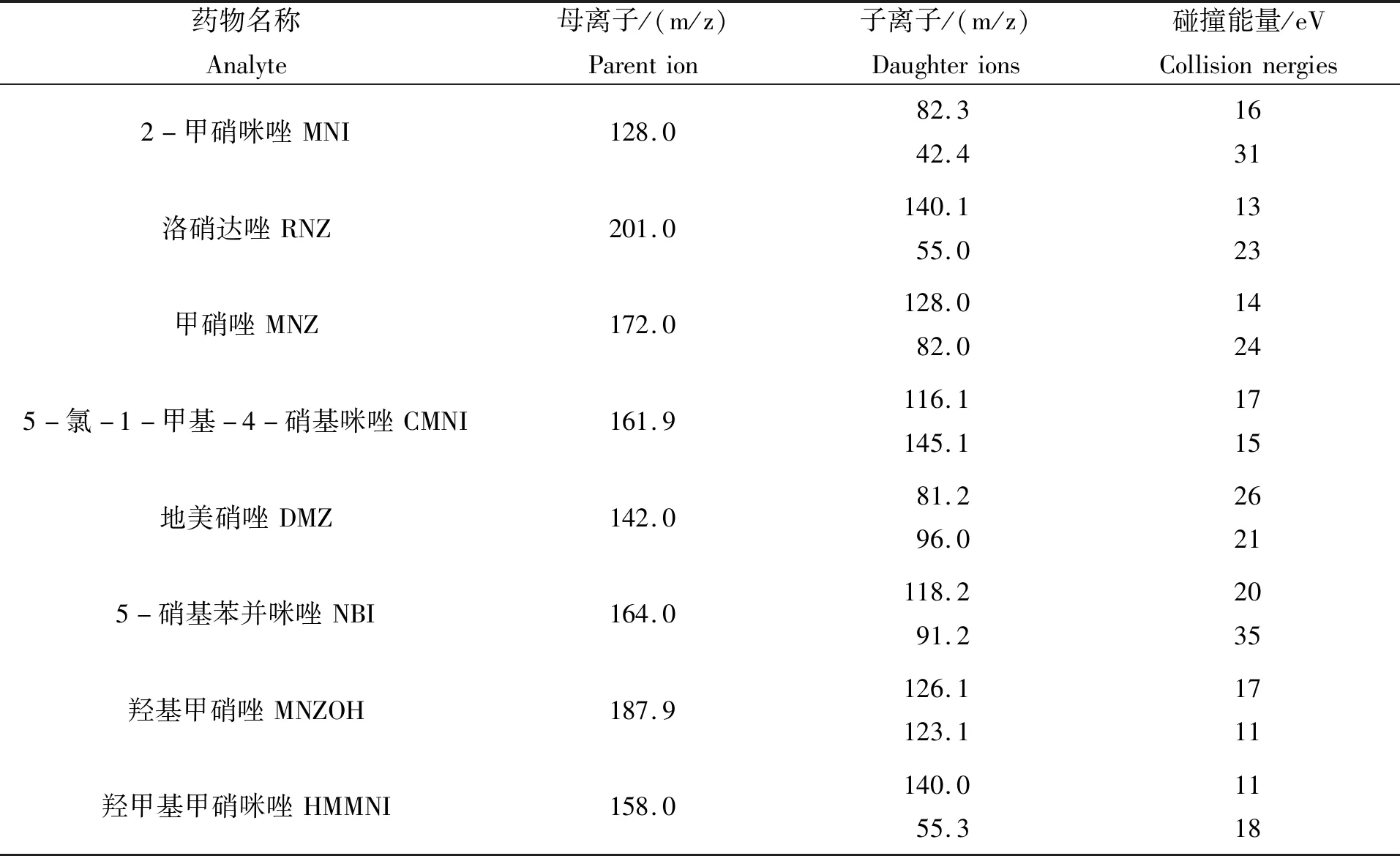
续表2
1.5 标准曲线绘制
准确称取适量的每种硝基咪唑类药物标准品,先用甲醇配制成浓度为1.0 mg/mL标准储备液。再用甲醇稀释成浓度为1.0 μg/mL硝基咪唑及其代谢物标准中间液。
根据每种硝基咪唑及其代谢物的灵敏度和线性范围,分别吸取浓度为1.0 μg/mL硝基咪唑标准中间液5.00~200 μL,配制成浓度为5.00、10.0、20.0、50.0、100、200 ng/mL系列混合标准工作液,吸取10.0 μL进样分析,以各组分的峰面积(Y)和相对应的质量浓度(X)进行线性回归,绘制标准曲线。
1.6 回收率与精密度
称取(5.00±0.05)g匀浆后的样品,分别加入一定量不同浓度的硝基咪唑类药物标准溶液,然后按同样的样品前处理方法进行提取、净化以及高效液相色谱-串联质谱检测,计算各种硝基咪唑药物的回收率及相对标准偏差。
2 结果与分析
2.1 检测方法的确定
2.1.1 质谱条件的优化
实验采用蠕动泵,分别将10种硝基咪唑类药物标准溶液(质量浓度均为1 μg/mL)以10 μL/min的流速注入电喷雾离子源,在ESI正离子模式下,对10种硝基咪唑药物的质谱参数进行优化。选择离子丰度最强的子离子作为定量离子,第二强的子离子作为定性离子。最后在选择反应监测正离子模式下,再次优化碰撞能量等质谱参数。优化后硝基咪唑类药物及其代谢物的质谱参数见表2,总离子流图见图1。
2.1.2 色谱条件的优化
硝基咪唑类药物及其代谢物是一种中等极性化合物,已有文献证实反相色谱柱更适合该类化合物的分离与检测[18]。因此,实验评估了Thermo Hypersil Gold C18柱和 Agilent ZORBAX SB-C18柱的分离效果,发现10种硝基咪唑类药物在两种色谱柱上均可实现较好的分离。但是使用Agilent ZORBAX SB-C18柱分离时,色谱峰拖尾,而采用 Thermo Hypersil Gold C18柱作为分析柱时,基线稳定,色谱峰拖尾较少,峰形更好。因此,实验采用Thermo Hypersil Gold C18色谱柱作为分析柱。
由于硝基咪唑类药物易溶于甲醇和乙腈,微溶于水,实验选择反相色谱常用的甲醇-水和乙腈-水两种流动相体系,并在水相中加入0.1%甲酸促进离子化,研究其对分离效果和峰形的影响。结果发现,采用0.1%甲酸水溶液-乙腈体系作为流动相时,10种硝基咪唑类化合物的分离效果较差,而0.1%甲酸水溶液-甲醇作为流动相时,可以更好地提高检测的灵敏度和分离效果,获得理想的色谱峰。本文还研究了在0.1%甲酸水溶液中加入5.0 mmol/L乙酸铵对10种硝基咪唑类药物分离效果的影响。结果显示,当在流动相中加入一定量的乙酸铵后,部分目标物响应强度下降,而且色谱峰出现峰展宽的现象。因此本实验选用甲酸水溶液-甲醇作为流动相。此时目标化合物能被较好地分离,其色谱图见图1。
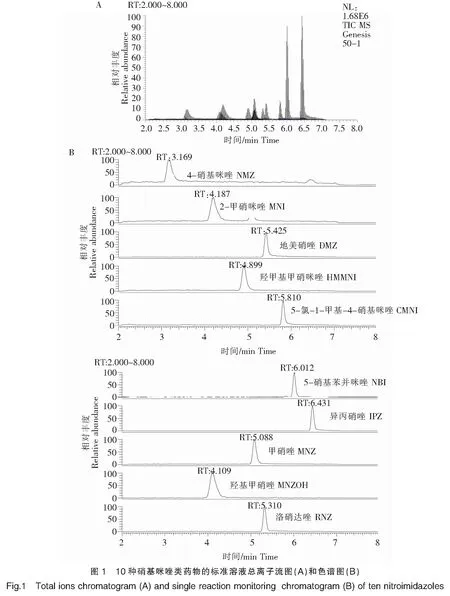
综上所述,本研究选用Thermo Hypersil Gold C18柱、0.1%甲酸水溶液-甲醇作为流动相对硝基咪唑药物进行分离分析。
2.2 前处理方法的选择
常用于提取动物组织内的硝基咪唑类药物的溶剂主要有甲醇、乙腈、乙酸乙酯等[18-20],乙腈和甲醇虽然能提取硝基咪唑及其代谢物,而且能沉淀蛋白质,但是其也能与水混溶,会导致水溶性杂质较多,给后续的净化与分析带来干扰,加标回收率比较低;相比甲醇和乙腈,乙酸乙酯极性较低,在提取硝基咪唑类药物的同时能减少水溶性杂质的溶出;而且硝基咪唑类药物有一定的热敏感性,如果在旋转蒸发步骤中水浴温度过高,将导致目标物的损失,因此在实验过程中应尽可能选用沸点较低的有机溶剂,乙酸乙酯低沸点的特点可以弥补在旋转蒸发步骤中过高的水浴温度或过长的浓缩时间导致回收率偏低的缺点,更适合作为提取溶剂。因此实验首先选用乙酸乙酯进行考察。因动物源性食品样品含有大量的脂肪等杂质,提取液用正己烷脱脂。研究发现,乙酸乙酯溶剂对10种硝基咪唑类化合物的回收率可达70.0%~120%。考虑到硝基咪唑类药物具有酸碱两性性质,即在弱碱性条件下呈游离分子状态,在弱酸性状态下呈质子化状态,因此提前在样品中加入磷酸二氢钠缓冲液(pH=8.0)预处理,使目标物以游离分子状态存在,再加入乙酸乙酯进行提取。经比较发现,加入pH=8.0的弱碱性磷酸盐缓冲溶液时,样品回收率在70.0%~120%范围内,且平行性比在中性状态下提取的更好。
2.3 基质效应评估
由于水产品样品基质复杂,常对待测物产生一定的基质效应。因此本研究用流动相和空白水产品样品经提取后的定容液分别绘制系列浓度的硝基咪唑标准曲线,将两种方法配制标准曲线的测定结果进行比较,即基质效应(%)=空白样品提取液配制标准曲线的测定结果/流动相配制标准曲线的测定结果×100,以此评价样品所产生的基质效应。结果显示,在样品基质中各种硝基咪唑类药物的基质效应在86.8%~108%之间,结果见图2。可见,经本方法进行前处理后10种硝基咪唑类药物的基质效应均在可接受的范围内。

2.4 线性关系和检出限
在上述优化的色谱和质谱条件下,测得标准溶液色谱图(图1)。从图中可知,在该优化条件下,各种硝基咪唑类药物得到了很好的分离,且未发现杂质色谱峰的干扰。同时对序列浓度的标准溶液进行测定,以峰面积对硝基咪唑类药物的质量浓度作线性回归,得标准曲线(表3)。试验结果表明,10种硝基咪唑类药物线性关系良好,线性相关系数均大于0.995 0,说明该方法适用于硝基咪唑类药物的定量分析。
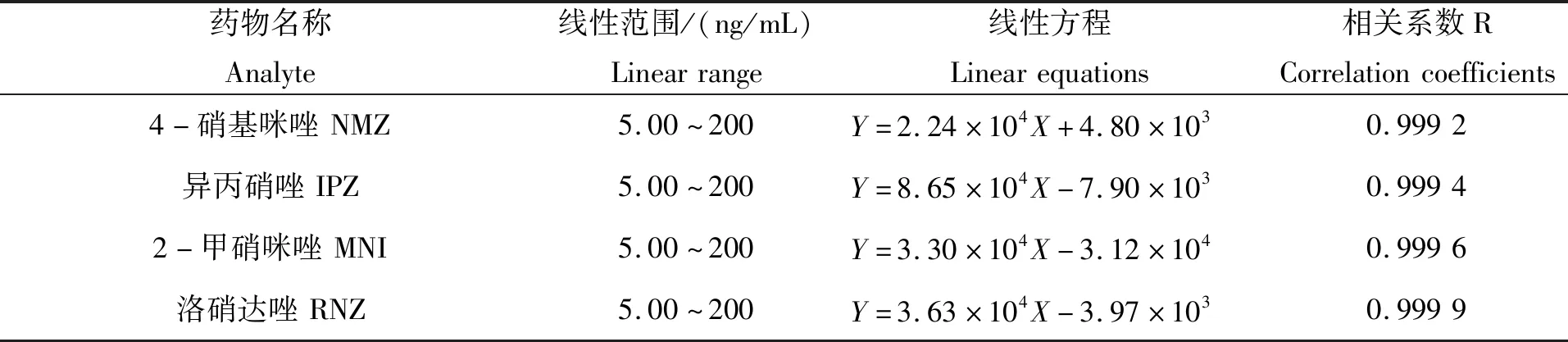
表3 10种硝基咪唑类药物的线性方程及相关系数Tab.3 Linear equations and correlation coefficients of ten nitroimidazoles
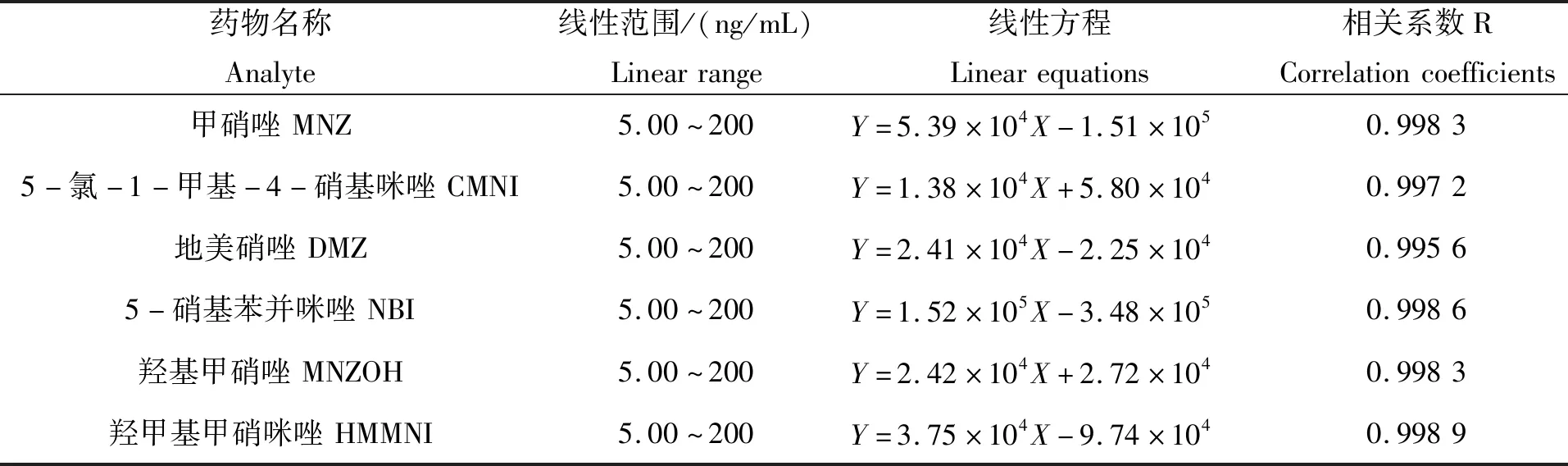
续表3
2.5 方法的灵敏度和回收率实验
由于在液质联用仪分析样品的过程中,存在基质效应,以空白样品进行加标处理后,在以信噪比(S/N)>3的前提下,求得10种硝基咪唑类药物的检出限为1.00 μg/kg。以不含硝基咪唑类药物的空白样品为研究对象,本实验以鱼肉为例,在样品中分别加入1.00、2.00和5.00 μg/kg三个浓度水平的硝基咪唑类标准品,每个浓度做三个平行实验,以考察方法的准确度和重现性,图3为空白样品和添加量为5.00 μg/kg的加标样总离子流图,加标回收率和精密度的实验结果见表4。可知,在不同的加标浓度下,样品的回收率均在70.2%~119.0%之间,相对标准偏均差(n=3)在13.5%以下。从试验结果可以看出,该方法的准确度和精密度满足水产品中硝基咪唑类药物残留量检测的要求。

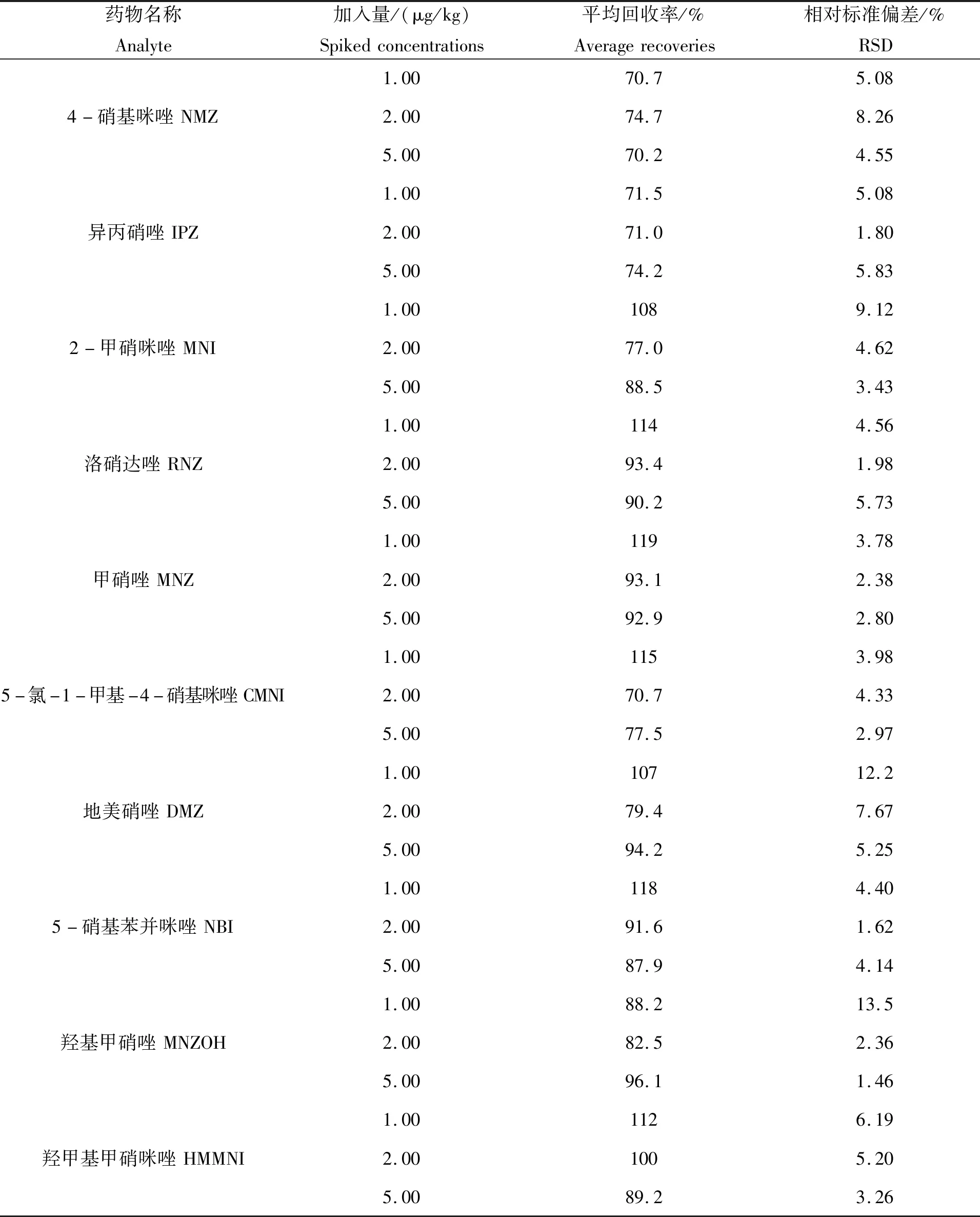
表4 鱼肉加标样的回收率和相对标准偏差Tab.4 Recoveries and RSDs for spiked fishes sample
2.6 实际样品测定
采用本方法对本地区市售的鲤鱼、草鱼和鲫鱼等共88份样品进行了检测,均未检出硝基咪唑类药物。相关部门应继续加强水产品中硝基咪唑药类物残留量的监测,保障水产品的质量安全。
3 结论
本研究在相关文献和标准的基础上,采用高效液相色谱-串联质谱法,通过对前处理条件和检测条件的优化,实现水产品中10种硝基咪唑类药物残留的同时测定。实验采用乙酸乙酯提取水产品中的硝基咪唑并用正己烷除脂,净化后在液相色谱串联质谱仪上检测,以甲酸水溶液/甲醇作为流动相进行梯度洗脱,改善了硝基咪唑类药物的响应值和峰形,提高检测的灵敏度。
在本检测条件下,10种硝基咪唑线性范围为5.00~200 ng/mL。线性相关系数均大于0.995 0。对鱼肉空白样品进行10种硝基咪唑的加标实验,加标量分别为1.00、2.00、5.00 μg/kg,回收率在70.2%~119%之间,相对标准偏均差(n=3)在13.5%以下。该检测方法与原有的标准方法[20]相比,操作更简便,灵敏度更高,适用于水产品中硝基咪唑类药物的大批量检测。
猜你喜欢
粘接(2022年8期)2022-08-19
导弹与航天运载技术(2022年2期)2022-05-09
现代仪器与医疗(2022年1期)2022-04-19
小作家报·教研博览(2022年11期)2022-04-02
分析化学(2019年3期)2019-03-30
分析化学(2018年12期)2018-01-22
食品界(2017年7期)2017-08-24
科技视界(2016年26期)2016-12-17
价值工程(2016年29期)2016-11-14
当代化工(2016年3期)2016-07-10
