
脑小血管疾病合并中枢神经系统退行性疾病机制的研究进展
2022-02-19 08:54武冬冬陈玉辉刘芳刘银红蒋景文
诊断学(理论与实践) 2022年5期
武冬冬,陈玉辉,刘芳,刘银红,蒋景文
(北京医院保健医疗部神经内科 国家老年医学中心中国医学科学院老年医学研究院,北京 100730)
脑小血管疾病(cerebral small vessel disease,CSVD)是指各种病因影响脑内小动脉及其远端分支、微动脉、毛细血管、微静脉和小静脉所导致的一系列临床、影像、病理综合征[1]。典型的CSVD 病变包括脑白质高信号、腔隙性脑梗死和脑微出血,其他影像学特征还包括皮层表面铁沉积、血管周围间隙和皮层下小梗死[2]。CSVD 患者可无临床症状,但随着其疾病负担逐渐加重,患者可出现认知障碍、情感障碍、步态障碍等症状[3],往往类似于中枢神经系统退行性疾病(简称神经退行性疾病)的临床表现。CSVD 在成人中很常见,与神经退行性疾病如阿尔茨海默病(Alzheimer′s disease,AD)、帕金森病(Parkinson′s disease,PD)和额颞叶变性(frontotemporal lobar degeneration,FTLD)等常合并出现。已有越来越多的证据表明,CSVD 可能在神经退行性疾病的神经生物学中发挥重要作用,且CSVD 对药物治疗神经退行性疾病疗效的影响仍然是一个有争议的话题。因此,关注CSVD 对神经退行性疾病的潜在作用和临床影响十分有意义。本文总结了CSVD 合并神经退行性疾病共同的分子机制和病理生理学,分析CSVD 对常见神经退行性疾病(AD、PD、FTLD)的潜在作用,总结CSVD 合并神经退行性疾病的生物和分子影像标志物以及预测治疗领域未来可能的研究方向。
CSVD 合并神经退行性疾病共同的发病分子机制
AD、PD、FTLD 等神经退行性疾病在临床表现上虽有显著差异,却具有共同的病理生理基础,包括蛋白质错误折叠、朊病毒样增殖和蛋白质聚集物的逐渐积累[4]。在神经退行性变的进程中同样会涉及到CSVD 的多种相关机制(见图1)。
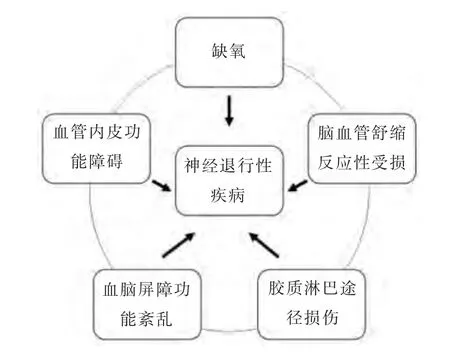
图1 多种CSVD 相关机制参与神经退行性变过程的示意图
一、缺氧
缺氧是CSVD 的主要发病机制之一,而在CSVD 合并神经退行性疾病发生机制中,缺氧相关研究较多。缺氧会导致神经元淀粉样变和tau 蛋白病,而这两者是AD 的病理特征。缺氧诱导因子1α 可使参与淀粉样蛋白级联反应的β-分泌酶和γ-分泌酶的转录增强,下调Aβ 降解酶(即脑啡肽酶)[5],导致Aβ 蛋白在神经炎性斑沉积。在啮齿动物中,缺氧通过丝裂原活化蛋白激酶的介导,引起神经元中过度磷酸化的tau 蛋白积累,随后形成异常的tau 蛋白丝,类似于人类tau 蛋白病中的tau 蛋白丝[6]。另有研究发现,TAR DNA 结合蛋白43(TDP-43)以及与FTLD 相关的融合肉瘤是缺氧时最易聚集的蛋白质,提示缺氧是神经退行性疾病中蛋白质错误折叠和聚集的触发条件[7]。
二、血管内皮功能障碍
内皮功能障碍在CSVD 与神经退行性疾病的联系中起着关键作用。缺氧会导致线粒体机制紊乱,抑制星形胶质细胞对谷氨酸的再摄取,引起谷氨酸介导的毒性反应,改变血管特异性基因的表达,从而导致血管内皮功能障碍[8]。在中枢神经系统中,内皮细胞的功能包括对血管张力和血流的调节,防止血栓、炎症和纤维化形成,免疫系统调节,跨越血脑屏障(blood-brain barrier,BBB)的分子的运输,以及细胞毒性产物的清除。研究已证实,PD 的内皮功能受累,在使用MPTP 治疗的动物模型和PD 患者的脑样本中,均检测到位于黑质、脑干和大脑皮层的毛细血管缺失和异常的碎片状毛细血管[8]。
内皮相关信号传递是由一系列血管活性物质介导的,一氧化氮(NO)是其中之一,它也参与线粒体的功能活动、突触传递、维持神经元和神经胶质的稳态[9]。内皮功能障碍会引起NO 可利用性降低,导致血管舒张受损,局部脑血流量减少,氧化应激加重。NO 是由一氧化氮合酶(nitric oxide synthase,NOS)催化合成的,NOS 有3 种同工酶。有证据表明,在培养的脑微血管内皮细胞中内皮型NOS(eNOS,NOS 的3 种同工酶之一)失活,进而引起淀粉样前体蛋白和淀粉样前体蛋白裂解酶1 的表达增加,最终导致Aβ 增加[10]。
三、脑血管舒缩反应性受损
内皮功能障碍也是脑血管舒缩反应性(cerebral vasomotor reactivity,CVR)受损的原因。CVR 是指脑内小动脉或毛细血管响应各种收缩或舒张刺激时脑血流的变化量,反映的是脑血管储备和调节的能力。临床研究表明,AD 与PD 中均存在CVR 受损,且与轻度认知障碍(mild cognitive impairment,MCI)向痴呆发展相关[11-12]。AD 患者的淀粉样蛋白沉积不仅累及脑实质,还累及颅内血管壁,引起动脉收缩,造成CVR 受损。此外,这些淀粉样蛋白沉积物会损害内皮细胞产生NO 等血管扩张剂的能力,导致血管舒张受损,进而影响CVR 和其他调节脑血流量的机制,如自动调节机制和神经血管耦合机制[13]。来源于Meynert 核的胆碱能投射,可直接通过释放乙酰胆碱,或间接通过刺激脑干内的中间神经元释放NO 来诱导血管舒张。因此,AD 和PD 中常见到的胆碱能传递功能障碍,是导致CVR 受损的另一机制[14-15]。
四、BBB 功能紊乱
神经血管单元由血管内皮细胞、周细胞、血管平滑肌细胞、星形胶质细胞和神经元组成,是BBB 功能完整的基础,而内皮功能障碍会对神经血管单元产生不利影响。当BBB完整性破坏时,血管周围间隙的Aβ 引流减少,造成Aβ 在脑内异常堆积[16]。脑内Aβ 的稳态需要通过内皮细胞表达的2 种受体相互作用而调节,其中脂蛋白受体相关蛋白1负责从大脑中清除Aβ,而晚期糖基化终末产物受体则介导Aβ 从循环中重新进入大脑。有证据表明,在AD中,晚期糖基化终末产物受体上调,脂蛋白受体相关蛋白1 下调[16]。载脂蛋白E(apolipoprotein,ApoE)与血管周围间隙Aβ 的相互作用可能与淀粉样肽的血管周围引流有关,进而影响内皮功能[16]。APOE 基因多态性因其等位基因与Aβ 相互作用的差异,而引起该引流途径强度的差异。在CSVD 的血管壁病变中同样检测到APOE[17],其等位基因ε4 被认为是散发性AD 的主要遗传危险因素,也与CSVD 的严重程度相关[18]。
五、胶质淋巴途径损伤
内皮功能障碍也可能与胶质淋巴途径的损伤有关,胶质淋巴途径是近期在啮齿类动物大脑中发现的一种液体清除系统,其通过水通道蛋白4 将有毒溶质从大脑清除到脑膜和颈部淋巴引流血管。tau 蛋白和淀粉样蛋白的清除率下降会进一步加重它们在脑内的沉积,进而加重AD 进程;而CSVD 小动脉收缩导致血管顺应性降低和血管周围间隙扩张,引起动力源不足,使胶质淋巴途径对有毒溶质的清除率降低,又会进一步加重CSVD 进程。研究已证实,在AD 和CSVD 的动物模型中存在胶质淋巴功能障碍,这可能与水通道蛋白4 表达紊乱有关[19]。
可见,CSVD 相关的多种发病机制参与了神经退行性变的过程(见图1),在神经退行性疾病的发生、发展中有着重要的意义,其中可能还涉及到其他更多的机制,目前缺乏具体的实验数据,尚未阐述清楚,有待进一步研究。
CSVD 合并常见神经退行性疾病的病理机制及表型
一、CSVD 与AD 的病理及表型关系
AD 是最常见的导致痴呆的神经退行性疾病[20],而CSVD也可导致痴呆。有证据表明,高血压、糖尿病、血脂异常、高同型半胱氨酸血症和ε4 等位基因是AD 合并CSVD 的危险因素[21]。AD 常呈现AD 与CSVD 混合的病理表现,且随年龄增加,混合病理共存概率增加[22],高达84%的老年AD 患者合并CVD 的病理改变。而这2 种病理类型对认知障碍的贡献权重需要更进一步的研究。
1.CSVD 与AD 发展的关系:值得研究的问题之一是CSVD 是否在AD 整个连续发展过程中均发挥作用,即从临床前阶段到前驱期阶段、从MCI 到痴呆。近期一项针对临床前AD 的纵向调查显示,CSVD 及AD 的病理改变在很大程度上对MCI 进展的风险具有独立的影响,而非协同作用[23]。关于CSVD 对AD 发展的影响,不同研究得出了不同的结果。在AD 神经成像初步研究中,基线及1 年随访时,发现较大的脑白质高信号(white matter hyperintensities,WMH)体积与更严重的认知损害相关[24]。而在另一项来自牛津的研究记忆和衰老项目研究,在161 例经病理证实的AD 患者中,脑皮层下CSVD 严重程度与患者生命最后2 年的认知检查评分无相关性,而与年龄显著相关,且女性的严重程度略高于男性[25]。
2.AD 与潜在的CSVD 病理改变:另一个值得研究的问题,AD 患者是否合并潜在的CSVD 的病理特征。研究发现晚发型AD 患者(发病年龄大于70 岁)比早发型(发病年龄小于60 岁),存在更多见的WMH 和腔隙性梗死[26],且晚发型AD 与CSVD 之间的这种关联可能并不是由AD 的遗传易感性(ε4 等位基因和AD 家族史)导致。在PREVENT 痴呆研究中,在入组的40~59 岁认知正常受试者中,研究者未发现ε4 携带者或痴呆家族史与CSVD(WMH 体积和微出血)之间存在关联[27];相反,在另一组平均年龄为58 岁的认知正常的受试者中,研究发现WMH 分布与ε2 等位基因相关[28]。在MCI 和痴呆患者中发现,相对于ε4 携带者,ε2 携带者有更大的WMH 体积(平均10.4 mL 比7.3 mL),且比ε3 纯合子携带者具有更高的脑微出血发生率(37.5%比18.3%)[29]。虽然ε2 携带状态在AD 患者中并不常见,曾被认为是AD的保护因素,但ε2 携带状态的AD 相比ε4 携带者及ε3 纯合子而言,具有特殊的临床表征,即CSVD 负担更大、非遗忘障碍更明显、脑萎缩分布不对称(左侧大于右侧)以及颞叶内侧结构相对保留[29]。
Ferreira 等[30]研究了CSVD 在AD 患者中的患病率和分布状况,根据MRI 上的脑萎缩模式将AD 分为经典型(海马及联络皮质均衡萎缩)、边缘系统优势型(海马普遍萎缩)、海马保留型(主要为联络皮质萎缩)和轻度萎缩型4 种不同亚型。研究得出,WMH 和微出血的发生率在边缘系统优势型AD 中最高,在轻度萎缩型中最低。各亚型均表现出在额叶和脑后区域更明显分布的WMH。此外,微出血部位的分布状况在不同亚型间存在差异,经典型和边缘系统优势型在所有脑叶区均有较高的CSVD 发生率,海马保留型在后叶、小脑和深部有较高的CSVD 患病率,轻度萎缩型只在后叶和小脑有较高的CSVD 患病率。
二、CSVD 与PD 表型的关系
在原发性PD中,近25%的患者在病程中会合并脑血管疾病[31]。临床、影像和病理学研究表明,CSVD 及CSVD 危险因素如糖尿病、高血压和血脂异常等对PD 患者的认知以及运动功能,特别是姿势和步态均有负面影响[32],即使是亚临床的CSVD 也与PD 更严重的运动障碍及步态障碍有关[33]。除已知的与α 突触核蛋白相关的神经退行性变和脑淀粉样变是PD 认知障碍进展的原因外,CSVD 负担已被证明是另一相关因素[33]。此外,Hoehn-Yahr 分期与CSVD 之间也存在显著正相关,即CSVD 负担是PD 疾病进展的相关因素[31]。
PD 的脑血管损伤可能对PD 疾病的不同表型产生不同的影响。已有研究表明,PD 患者的CSVD 负担与步态、姿势障碍有关,与年龄、高血压、缺血性卒中史、低密度脂蛋白和血胆固醇水平以及简易精神状态检查量表和蒙特利尔认知评估量表评分无关[32]。脑白质病变的负荷会加重PD 的躯干中轴障碍和运动迟缓,但并不影响震颤或强直[32]。最近的一项多中心研究证实,CSVD 会加重PD 的症状,且对左旋多巴长期治疗的反应产生负面影响。PD 和CSVD 疾病叠加可能会产生混合的运动表型,部分患者的运动和认知障碍对抗PD 治疗的疗效不佳可能与合并CSVD 有关[32]。
三、CSVD 与FTLD 病理及表型关系
FTLD 代表一组具有异质性的神经退行性疾病,其病理特征是不同蛋白(如TDP-43、融合肉瘤蛋白等)的聚集,患者的临床表现包括痴呆、进行性精神行为异常、原发性进行性失语、帕金森症和运动神经元-相关表型[34]。以tau 蛋白(FTLD-tau)为特征的FTLD 亚型包括Pick 病、进行性核上性麻痹和皮质基底节变性。如前所述,缺氧是蛋白质错误折叠和聚集的触发因素,也会出现在非AD 相关的tau 蛋白病中。因此,CSVD 是否在所有FTLD 亚型中发挥类似的作用尚待进一步研究。
在一项尸检研究中发现,Pick 病比其他FTLD-tau 亚型表现出更高的CSVD 负担,还发现CSVD 样病损区域与额颞叶皮层中tau 相关的脱髓鞘区域存在交叉。此项研究中Pick病患者发病年龄最年轻(平均发病年龄约为57 岁),提示CSVD 可能导致具有潜在Pick 病病理的患者临床更早发病,然而类似程度的CSVD 与该研究中的老年组患者脑中Pick病相关的脱髓鞘发生并无关联[35]。这一发现提示,FTLD-tau的临床表现是由Pick 病中tau 沉积于皮层所致,可能并非CSVD 的病理作用。关于CSVD 与FTLD 之间的关系尚待进一步研究。
CSVD 合并神经退行性疾病的标志物
头颅MRI 是诊断CSVD 的首选影像学方法[36],但CSVD患者存在相关神经影像学发现,可能在临床上并无症状,或出现类似于神经退行性疾病的临床表现,因此单靠脑影像表现并不能确定CSVD 是否足以解释临床表现及确定是否同时需要考虑合并神经退行性疾病。此外,当CSVD 过于严重和广泛时,影像学表现仅能代表不同疾病病理叠加的最终图像,不能体现出不同疾病的阶段及其分子机制。近年来,关于疾病生物标志物的研究被寄予厚望。在临床症状的基础上,结合CSVD 和AD、PD 等神经退行性疾病的生物标志物,如脑脊液生物标志物和分子影像学标志物,可以极大提高疾病诊断的特异度和灵敏度,并能很好地反映出疾病的进展程度。
一、CSVD 与AD 的脑脊液生物标志物
脑脊液是中枢神经系统疾病生物标志物的可靠来源。结合脑脊液中反映淀粉样变的A β42/A β40 比值,和反映tau蛋白病的苏氨酸181 磷酸化的tau 蛋白(p-tau),就可以证明AD 相关神经生物学变化的存在,从而确诊AD[20]。CSVD 的脑脊液也具有某些特征。研究发现,由少突细胞来源的髓鞘相关分子,如髓鞘碱性蛋白,反映了髓鞘再生过程,髓鞘碱性蛋白水平的升高与CSVD 的临床表现有关,并可与AD 相鉴别[37],有望成为CSVD 的脑脊液生物标志物。另一个标志物神经丝轻链(neurofilament light chain,NfL)可反映轴突受损,在CSVD 患者的脑脊液中很早就被发现升高,甚至在伴有白质改变的非残疾患者以及后期发展为血管性痴呆的患者在其MCI 阶段时就有增加[38]。如果脑脊液t-tau 反映的是受淀粉样蛋白影响的神经元分泌tau 蛋白的增加,那么脑脊液NfL 反映的则是神经元的受损,不受淀粉样蛋白影响[39]。因此,与单纯AD 相比,在AD 伴CSVD 的病例中脑脊液NfL水平会更高。NfL 水平对判断在神经退行性疾病中是否合并CSVD 具有辅助作用,未来可在除AD 外的其他神经退行性疾病中开展进一步研究。
对CSVD 相关痴呆患者(包括单纯CSVD 和CSVD/AD混合病理)的研究显示,相对于单纯AD 患者及健康对照者而言,此类患者脑脊液中的α1 抗胰蛋白酶、组织金属蛋白酶抑制剂1、纤溶酶原激活物抑制剂1 和载脂蛋白H 水平升高[40]。
结合不同的生物标志物,包括反映AD(Aβ42、p-tau 和t-tau)、轴突损伤(NfL)、脱髓鞘的和基质重塑通路(组织金属蛋白酶抑制剂1 和基质金属蛋白酶)的,对于辅助鉴别CSVD 与AD[37]会有所帮助。
二、CSVD 与PD 的分子影像学标志物
脑网络是大脑结构和功能的映射,大脑的高级认知功能与脑网络密切相关。传统的MRI 标志物注重局部脑区损伤,脑网络构建更注重对不同结构损伤带来的改变进行全脑信息整合。脑网络分为结构性脑网络和功能性脑网络。MRI 弥散张量成像(diffusion tensor imaging,DTI)可无创追踪脑白质纤维并反映其解剖连通性,结构性脑网络的完整性可以通过DTI 进行定量评估。静息态功能磁共振是构建功能性脑网络的主要影像技术[41]。
1.CSVD:目前已有较多研究应用DTI 技术,验证了脑白质微结构损伤与CSVD 常见的临床表现(如认知障碍)显著相关。Tuladhar 等[42]构建了436 例CSVD 患者的DTI 结构性脑网络,随访5 年后有32 例新发痴呆,而这些患者在基线时就已经表现出了脑网络属性的异常(包括结构网络强度、网络整体效率以及局部效率的降低),提示脑网络的破坏对于CSVD 认知功能障碍的发生、发展至关重要。
2.神经退行性疾病:在神经退行性疾病中,研究者发现了脑网络功能连接的异常。Baggio 等[43]在一项采用静息态血氧水平依赖的MRI 研究中发现,PD 合并MCI 患者的背侧注意网络[又称视空间注意网络,负责管理空间注意和视觉运动,参与目标导向的自上而下(内源性)的注意定向]和右侧额叶区域之间的连接性降低,与注意力、执行功能的受损具有明显相关性。默认网络(default mode network)是指大脑在无外加任务时自发脑功能活动相对活跃的区域,该研究还发现默认网络与枕叶及顶叶区域的连通性增加,与其视觉空间、视觉感知表现更差具有相关性,提示PD-MCI 患者网络的差异连通性变化可能是PD 患者发生不同临床特征认知障碍的病理生理学基础。功能连接异常在CSVD 中也被认为可能与认知功能障碍相关[44]。已有研究认为,默认网络连接改变可能是导致CSVD 认知功能减退的关键。可见,CSVD与神经退行性疾病均可通过改变脑网络而引起认知障碍,但两者的具体机制有何不同尚不清楚。脑网络研究已成为近年的研究热点,将结构性脑网络和功能性脑网络进行整合的复杂网络分析,将为研究者提供更全面的视角,来探索CSVD 合并神经退行性疾病发生、发展的机制。
展望
CSVD 的发病率与年龄呈正相关,常与神经退行性疾病并存。脑血管病相关危险因素、CSVD 及中枢神经系统疾病之间的相互作用可能导致神经退行性疾病进程的加速。减少缺血引起的神经元损伤,使得共病的脑小血管病对运动和认知表现方面的损害降低,是治疗神经退行性疾病的关键。因此,在神经退行性疾病早期通过改善脑内血管重塑的治疗,以达到预防血管变性,为改善神经退行性疾病的运动和认知症状负担提供新的靶点。
既往对于某些神经保护剂是否对神经退行性疾病有疾病修饰作用的研究未得到阳性结果的原因之一,可能是忽略了CSVD 的潜在作用。迄今为止,大多数治疗神经退行性疾病的研究都集中在所谓的单效药物(single-action agents)的研发上,即通过逆转神经元功能障碍和(或)保护神经元,使神经元免受特定伤害。结果表明,单效药物并不能改变神经退行性疾病的病程。近年来,血管-神经元-炎症模型被提出,表明血管损伤、神经退行性变和神经炎症是发生于神经系统疾病病理生理的三联征。因此,与单效药物相比,作用于多个靶点的多效药物将有更多机会作用在神经退行性疾病复杂的疾病机制中[45]。一系列分子被认为是疾病修饰药物的潜在靶点,如NO[46]、血管内皮因子[47]等。血管-神经-炎症模型的研究前景广阔,有可能改变目前医学界对神经退行性疾病的认识,但开发新的神经与血管疗法,尚需进一步研究。
综上,在中老年人中,CSVD 常与神经退行性疾病同时发生。在共同的分子基础和神经生物学机制下,神经退行性疾病与CSVD 合并发生时,常会呈现出不同的临床表型、疾病进程和结局。反映淀粉样变、tau 蛋白病等神经变性的脑脊液生物标志物,可以与反映血管重塑、轴突损伤和脱髓鞘/髓鞘再生过程的脑脊液生物标志物相结合,并可采用分子影像学技术共同评估CSVD 与神经退行性疾病之间的联系。未来针对神经退行性疾病聚焦于血管-神经-炎症模型的新的治疗方法,会具有广阔的前景。
猜你喜欢
江苏卫生保健(2022年12期)2022-02-11
保健医苑(2021年7期)2021-08-13
中国毕业后医学教育(2020年5期)2020-12-06
中华肩肘外科电子杂志(2017年1期)2017-01-11
中华老年多器官疾病杂志(2016年9期)2016-04-28
中华老年多器官疾病杂志(2016年9期)2016-04-28
中外医疗(2015年5期)2016-01-04
医学研究杂志(2015年7期)2015-06-22
现代检验医学杂志(2015年6期)2015-02-06
现代检验医学杂志(2015年5期)2015-02-06
