
8 例髓鞘少突胶质细胞糖蛋白抗体相关疾病患者的临床和影像学表现分析并文献复习
2022-02-19 08:54林国珍任汝静
诊断学(理论与实践) 2022年5期
林 霞,高 超,黄 沛,王 刚,林国珍,任汝静
(1.甘肃省天水市第一人民医院神经内一科,甘肃 天水 741000;2.上海交通大学医学院附属瑞金医院a.神经内科,b.临床心理科,上海 200025)
髓鞘少突胶质细胞糖蛋白抗体相关疾病(antimyelin oligodendrocyte glycoprotein-IgG associated disorders,MOGAD)是一种免疫相关的,临床和影像学均表现为中枢神经系统炎性脱髓鞘的独立疾病。MOGAD 未统一命名前,曾被称为髓鞘少突胶质细胞糖蛋白(myelin oligodendrocyte glycoprotein,MOG)抗体自身免疫性脑炎[1]。MOGAD 可为急性单相病程或慢性复发病程,临床表现异质性大,以视神经炎、脑膜脑炎、脑干脑炎和脊髓炎表现为主[2]。MOGAD 患者对激素、免疫治疗敏感,大多呈复发缓解病程,预后良好,但部分患者可遗留不同程度的残疾。但也有少数影像学表现为颅内占位样病灶的MOGAD 患者,由于难以鉴别,给临床诊治带来困难。既往国内研究中对MOGAD 的报道以MOGAD相关的视神经炎、脑炎、脊髓炎等单一临床表型为主,而结合临床特征及影像学表现进行完整总结分析的较少,缺乏疗效及随访的分析。故本研究汇总了上海交通大学医学院附属瑞金医院神经内科诊断的8 例MOGAD 患者,分析其发病诱因、起病形式、临床表现以及血清学化验、脑脊液、脑电图、诱发电位及影像学表现,同时观察其治疗效果和预后,并结合相关文献进行综合探讨,以提高临床医师对MOGAD 的认知,为临床诊治提供参考。
资料与方法
一、资料
收集2018 年1 月至2021 年12月,上海交通大学医学院附属瑞金医院神经内科确诊的MOGAD患者的病历资料。所有患者均符合2020 年《抗髓鞘少突胶质细胞糖蛋白免疫球蛋白G 抗体相关疾病诊断和治疗中国专家共识》[2]中对MOGAD 的诊断标准。入组标准为符合以下所有条件。①血清和或脑脊液细胞法髓鞘少突胶质细胞糖蛋白(myelin oligodendrocyte glycoprotein,MOG)抗体阳性;②病灶累及中枢神经系统,临床表现包括视神经脊髓炎、脑膜脑炎、脑干脑炎、脊髓炎、瘤样脱髓鞘等,可为单一症状或以上症状的多种组合,这些症状的确认均有相应的影像学证据;③排除多发性硬化、视神经脊髓炎等脑白质脱髓鞘疾病;④所有患者均知情同意,且能配合本研究进行的随访工作。本研究的排除标准为存在以下任何一项,如有严重心、肺、肝、肾脏疾病的患者。
二、方法
最终有8 例MOGAD 患者被纳入本研究,收集其所有临床资料。①一般资料,包括发病诱因、起病形式、临床特征;②各项实验室检查结果,包括血常规、肝肾功、电解质、血糖、血脂、心肌酶、血沉、甲状腺功能五项、抗核抗体谱、肿瘤标志物等;③脊髓穿刺脑脊液检查结果,包括脑脊液压力、细胞数、蛋白质、葡萄糖、氯化物,脑脊液脱髓鞘抗体、寡克隆区带、自身免疫性脑炎抗体;④神经电生理检查结果,包括脑电图、诱发电位;⑤神经影像学检查结果,包括头颅MRI 平扫或增强、全脊柱MRI 平扫或增强检查;⑥治疗方案及预后、复发情况等。
1.MOG 抗体检测:采集所有患者的血液及脑脊液标本,送至上海金域医学检验所有限公司,用血清和或脑脊液细胞法进行MOG 抗体检测,血清抗体滴度≥1∶10 为阳性,脑脊液抗体滴度≥1∶1 为阳性。
2.头颅、脊柱MRI 检查:使用西门子公司、飞利浦公司3.0 超导型磁共振扫描仪进行扫描。头颅MRI 检查序列包括T1、T2、弥散加权成像、液体衰减反转恢复序列、表观弥散系数、T1 增强,脊柱MRI 序列包括T1、T2、T1 增强。
3.文献查询:在中国知网、万方医学网、维普数据库、PubMed 医学文献数据库中,分别以“髓鞘少突胶质细胞糖蛋白抗体相关疾病”“Myelin oligodendrocyte glycoprotein antibody-related diseases”“MOGAD”为检索词,查询1998 年至今国内外所有的相关文献,选择病例数大于20 的队列研究,排除其他疾病导致的MOGAD。通过文献复习对MOGAD 患者的临床特征、影像学特点、治疗进行综合分析。
结果
一、一般资料
8 例MOGAD 患者中男性6例,女性2 例;年龄为17~37岁,平均年龄为27.37 岁;病程为2 周~4 年。8 例患者中,亚急性起病2例,慢性起病6 例;1 例患者劳累为诱因,1 例感染为诱因,其余6 例无明显诱因。8 例患者发病前3 个月均无疫苗接种史。
二、临床表现
MOGAD 患者的临床表现多样,8 例患者中,4 例表现为肢体不同程度无力,3 例出现肢体抽搐,3 例出现视物模糊,复视、颜面部麻木、肢体麻木、发热、头痛、头晕各1 例。患者从出现症状到确诊的时间为2 周至4 年不等,首诊原因包括脊髓炎、脑炎、多发性硬化、颅内占位性病变。8 例患者临床表型如下,脑炎3例,脑干脑炎1例,视神经炎1例,长节段脊髓炎1例,炎性脱髓鞘假瘤2 例。
三、血清学、脑脊液检测
8 例MOGAD 患者血清学检测指标仅发现部分轻度异常,无特异性(见表1)。腰椎穿刺术脑脊液压力95~225 mmH2O,正常5 例(62.5%),增高3 例(37.5%)。细胞数(1~10)×106/L,正常3 例(37.5%),增高5 例(62.5%)。脑脊液糖、氯化物均在正常范围。脑脊液蛋白297.29~2 105.11 mg/L,正常4 例(50%),异常4 例(50%)。血液MOG 抗体滴度1∶1~1∶100 不等,脑脊液1∶10~1∶100 不等,血液、脑脊液双阳性患者6 例(75%),脑脊液MOG 抗体阴性2 例(12.5%),血液MOG 抗体阴性1 例(12.5%)。所有患者寡克隆区带、自身免疫性脑炎抗体以及中枢神经脱髓鞘抗体(包括抗AQP4 抗体、抗MBP 抗体、抗GFAP 抗体)均为阴性。

表1 MOGAD 患者血清学检测结果
四、神经电生理检查
8 例患者中,5 例进行了脑诱发电位检查,包括躯体感觉诱发电位、视觉诱发电位、脑干听觉诱发电位。结果显示5 例患者均存在单一或多个诱发电位异常,其中,躯体感觉诱发电位异常1例,视觉诱发电位异常1例,脑干听觉诱发电位和视觉诱发电位异常2例,躯体感觉诱发电位、脑干听觉诱发电位和视觉诱发电位异常1 例。4 例患者进行了脑电图检查,结果均出现异常,主要表现为慢波活动为主,偏侧性明显,伴尖波发放。
五、神经影像学检查
1.头颅MRI 检查:8 例患者中,7 例进行了头颅MRI 平扫检查,1 例正常,6 例异常,头颅MRI 提示颅内受累范围较广,表现为视神经、脑桥臂、丘脑、侧脑室旁、胼胝体、额叶、顶叶、枕叶、皮层下、小脑等多部位不同程度异常信号(见图1A~1E)。7 例患者均进行了头颅MRI 增强检查,其中,正常5例,异常2例,为脱髓鞘假瘤样表现,部分视神经、脑膜呈斑片、不规则强化。
2.脊髓MRI 检查:8 例患者中,6 例进行了脊髓MRI 检查,正常5例,异常1例,表现为颈髓内信号欠均匀,下段颈髓、胸髓内多发片状异常信号(见图1F)。
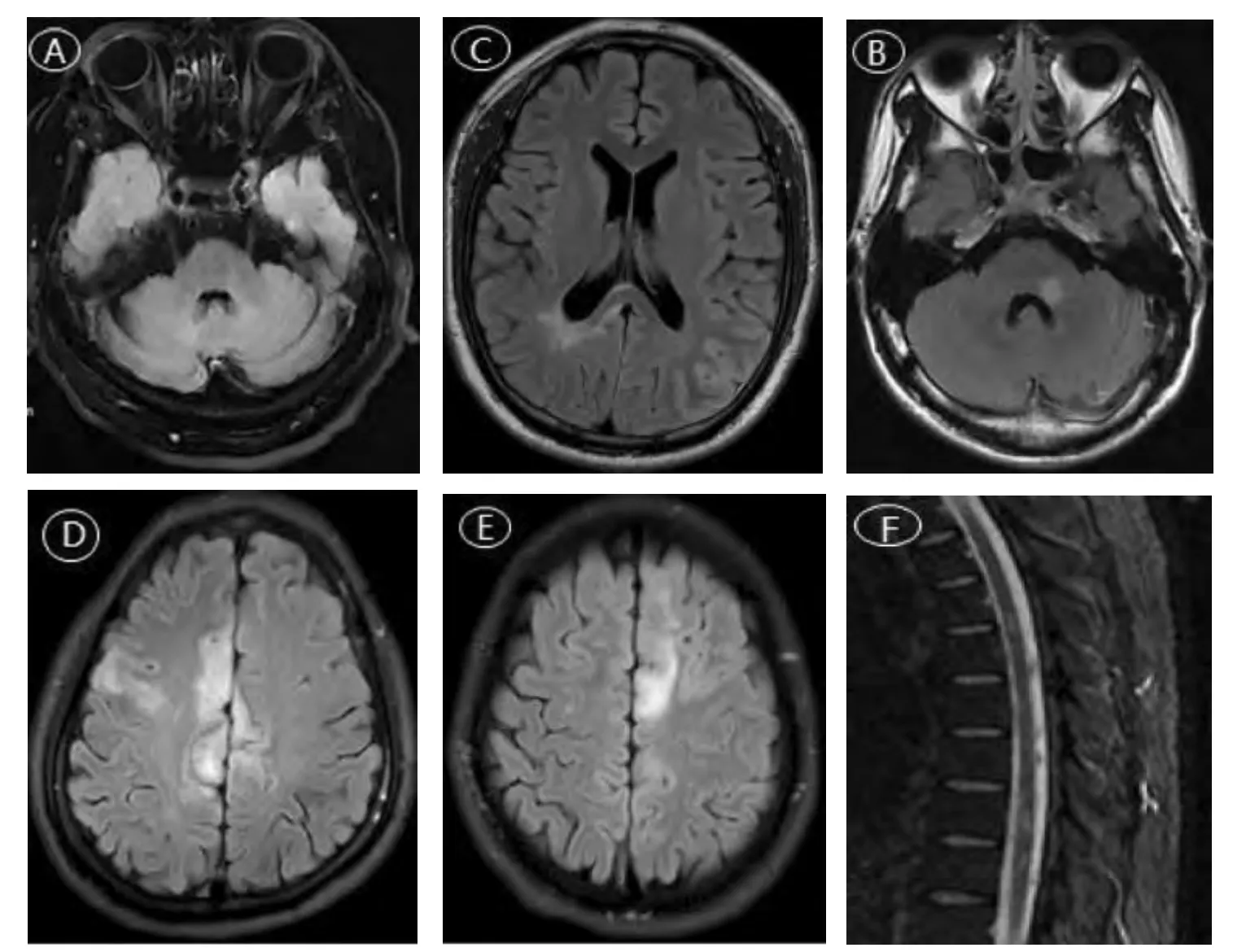
图1 MOGAD 患者影像学资料
六、治疗
8 例患者均使用大剂量甲泼尼龙冲击治疗,根据患者体重、病情严重程度选择剂量(500~1 000 mg/d),静脉滴注,5 d 后减至240 mg,5 d 后减至120 mg/d,再5 日后改为60 mg/d 口服,后根据病情缓慢减量至维持。有3 例患者还接受了联合丙种球蛋白0.4 g/(kg·d)治疗,连续5 日后停用;1 例患者联合口服环磷酰胺(50 mg/次,2 次/d)治疗。
七、随访
8 例患者随访2 个月~3年,其中7 例患者病情稳定,处于缓解期,无明显反复复发,定期随访中。1 例患者病情复杂,规律治疗仍复发,确诊MOGAD后,应用激素联合环磷酰胺免疫抑制治疗,患者病情趋于稳定,目前已门诊定期随访,至今3 年。患者再未出现发作性肢体无力抽搐、视物模糊、二便障碍等症状。
八、文献复习
检索国内外相关文献总计800 余篇,逐篇筛选了包括MOGAD 患者基本信息、临床特征、影像学表现、治疗、预后等项目的文献,剔除不完整病例及仅为单一临床表型的MOGAD 病例,共纳入资料完整的国内研究4项,国外研究1项,涉及病例数161 例(见表2)。综合分析相关文献结果提示,MOGAD 病例以青中年为主,成人发病年龄为18~67岁,男∶女=71∶90。患者的临床表型如下,63.98%的患者出现视神经受累,28.57%出现脑部受累,30.43%出现脊髓受累,10.56%出现急性播散性脑脊髓炎,无脑干脑炎表型。影像学表现提示视神经、全脑、脊髓均可受累。MOGAD 的治疗以糖皮质激素、免疫治疗为主,但患者的疾病容易反复,复发率为30.00%~43.39%,复发后患者对糖皮质激素、免疫治疗仍敏感,预后良好。

表2 MOGAD 相关文献总结[例(n)]
讨论
一、MOGAD 的临床表现
Cobo-Calvo 等[8]报道,MOGAD 患者1~71 岁各个年龄段均可发病,但儿童多发,而成人好发于30 岁左右,女性相较于男性更易发病。本研究报道的8 例MOGAD 患者中,以亚急性、慢性起病者多见,均为成人且青年居多,男女间的发病比例为3∶1。本研究报道的MOGAD 患者发病年龄与国内外相仿,但性别以男性更多见,这可能与病例数偏少有关,有待今后收集更多病例以验证。
MOGAD 是神经功能障碍的单相或复发过程,患者的临床表现多样,可出现中枢神经系统脱髓鞘疾病的所有临床表现,无特异性。MOGAD 的临床表型广泛,包括单侧和双侧前部视神经炎、长短横贯性脊髓炎、急性播散性脑脊髓炎、脑干脑炎、伴有或不伴有癫痫发作的皮质脑炎和炎性脱髓鞘假瘤等,且不同的临床表型可相互重叠[2]。既往文献报道显示,MOGAD 的临床表型在很大程度上取决于患者的年龄,儿童患者以急性播散性脑脊髓炎表型更多,而青少年和成人的视脊髓表型(视神经炎、脊髓炎和脑干脑炎)更多[9]。回顾文献报道的病例,国内MOGAD 患者的临床表型以视神经炎、脑炎、脊髓炎最为常见,而国外患者则以视神经炎、脊髓炎、播散性脑脊髓炎最常见。除此之外,还发现以自身免疫性脑炎、前庭神经元炎、菱脑炎患者出现的类固醇激素反应性慢性淋巴细胞性炎症伴脑桥血管周围强化症和极后区综合征等罕见的临床表型。本研究中8 例患者的临床表型分别为脑炎、脑干脑炎、视神经炎、长节段脊髓炎、炎性脱髓鞘假瘤等,其中炎性脱髓鞘假瘤是MOGAD 的罕见表型,其余表型与文献报道一致。
二、MOGAD 的实验室检查特征
MOGAD 诊断的金标准是血清和或脑脊液细胞法(CBA 法)MOG 抗体阳性。但部分患者的表现不典型,MOG 抗体检测结果可为阴性,而以中枢神经系统脱髓鞘疾病规律治疗后,其疾病仍反复复发。针对此类患者,反复进行多次血液、脑脊液MOG 抗体检测是必要的。本研究中1 例MOGAD 患者多次疾病复发,而在首次发病时其MOG 抗体检测结果为阴性,多次复发过程中最终检测出MOG 抗体而确诊。可见,临床医师对于部分MOG 抗体阴性,但脱髓鞘多次复发的患者,应定期复查脱髓鞘抗体,以尽早明确诊断。文献[8,10-11]报道显示,MOGAD 患者的血清、脑脊液均可检出MOG 抗体,脑脊液检出率较外周血清低,MOG 抗体滴度与疾病的严重程度、复发率、复发间隔时间均具有关联性。另有研究[8]发现,MOGAD 患者更高的MOG 抗体水平及持续血清抗体阳性与更高的疾病复发风险及残疾率增加有关。鉴于MOG 抗体在疾病复发时增加,而在治疗后转变为阴性,故认为在诊断时及整个MOGAD疾病过程中对该抗体进行定期监测是必要的,抗体水平降低可能表明疾病缓解。Jarius 等[13]建议,每6~12 个月对MOGAD 患者重新进行MOG 抗体检测可能会对治疗有所帮助。亦有相关文献报道显示,部分MOGAD 患者可合并寡克隆区带(oligoclonal band,OCB)阳性。本研究8 例患者均未合并OCB 阳性,提示MOGAD 鞘内免疫球蛋白合成相对少见,因本研究病例例数少,需后续继续观察。
三、MOGAD 的影像学表现
复习相关文献提示,MOGAD 的影像学表现与神经系统炎性脱髓鞘疾病重叠,中枢神经系统各部位均可受累,表现为视神经的增粗、肿胀,部分增强发现纵向强化。颅内病灶分布广泛,形态不一,无明显界限,部分呈斑点、条片状强化。脊髓MRI 表现为长或短节段性病灶,发现脊髓横断面“H”征,部分强化。本研究显示,MOGAD 患者的颅内病灶广泛,视神经、脑桥臂、丘脑、侧脑室旁、胼胝体、额叶、顶叶、枕叶、皮层下、小脑等多部位均可累及,但大脑灰质、皮层下U 型纤维无受累。MRI 图像上,MOGAD 脑部病灶呈斑片状,小病灶多见,边界不清,如表现为占位样,很少跨越中线,病灶通常为T1 低信号,T2、液体衰减反转恢复序列高信号,增强扫描可见不规则强化或不强化,部分脑膜、视神经可出现强化[14]。研究[15]发现,幕上深部白质、皮层或皮层下白质是MOGAD 最常见的受累部位,脑干受累亦相对常见,超过20%的MOGAD 患者存在丘脑或基底节受累,也有部分患者存在小脑、胼胝体不同程度受累,提示MOGAD 的颅内病变范围较广泛,与本研究患者的影像学表现一致。文献研究[16-17]发现,MOGAD 脊髓受累者出现长节段脊髓病变的比例较低,相应的其病灶的整体位置偏低。另有文献[18]总结发现,脊髓受累以纵向广泛的横贯性脊髓炎,伴有矢状T2 高信号髓内脊髓线、轴向“H”脊髓征(中央脊髓灰质T2 高信号)和脊髓圆锥受累为MOGAD 的特征性脊髓改变。而本研究仅有1 例以脊髓炎为表现的MOGAD 患者,其以长节段胸髓受累为主。以往的对采用影像学检查评估MOGAD 患者治疗后预后方面的研究较少,本研究8 例患者中7 例获得随访,患者经正规治疗后,复查影像学检查提示病灶范围均较前不同程度减少,其中2 例炎性脱髓鞘假瘤患者经积极、规范的治疗后,至复诊时其脱髓鞘假瘤样病灶均已消失,提示部分患者的MOGAD 可逆。
四、MOGAD 的治疗及预后
综合文献及本研究病例,MOGAD 的治疗与其他中枢神经系统脱髓鞘疾病类似,分为急性期治疗和缓解期治疗。MOGAD 患者对大剂量糖皮质激素冲击、免疫球蛋白、血浆置换等敏感,急性期应用疗效肯定,但容易反复,文献显示其复发率达30%~43.39%,且随着随访时间的延长,复发率呈逐年增高趋势,而随着患者复发次数的增多,其致残率也增高,故激素治疗需缓慢减量,小剂量维持,防止复发。对于部分经规范治疗后仍复发的患者,重复应用大剂量激素、免疫治疗仍有效,后期可考虑免疫抑制剂联合小剂量激素治疗,效果显著。文献[19-21]报道,激素和或免疫抑制剂长期维持,可有效降低MOGAD 的复发。亦有研究[22]发现,MOGAD 早期倾向于在中枢神经系统相同解剖位置复发,眼部受累最多。本研究报道的8 例患者经长程规范治疗后,7 例患者均长期缓解,仅1 例出现临床症状的复发,伴抗体滴度变化,但无相应的新发影像学病变出现,经联合环磷酰胺免疫抑制治疗后,其病情缓解。另有研究[23]探索了将干细胞治疗作为治疗罕见神经性自身免疫性疾病(包括MOGAD)的方法,随着对疾病认识的不断深入,漏诊率逐渐下降,未来研究可能会有更新的药物被逐渐应用到临床。
五、本研究的不足
本研究尚存在不足之处,限于MOGAD 病例罕见等客观原因,样本量少,上述病例仅体现了MOGAD 疾病的一部分特性,诸多的未知还需临床医师去不断的总结和发现,今后本团队将继续关注MOGAD 疾病的发展和变化,进一步提高对MOGAD的认知,为优化诊疗方案提供依据。
总之,MOGAD 亚急性、慢性起病多见,年轻患者居多,临床表现多样,表型广泛,脑脊液常规检验无特异性,影像学表现全脑、脊髓均可受累,患者对激素、免疫治疗敏感,治疗效果较好,预后较好,但易复发,需定期随访。MOG 抗体检测阳性是诊断MOGAD 的金标准,对疑似患者应尽早行MOG 抗体测定,必要时可多次检测,避免漏诊。
猜你喜欢
昆明医科大学学报(2021年4期)2021-07-23
医学与法学(2020年2期)2020-07-24
医学新知(2019年4期)2020-01-02
中国社区医师(2019年12期)2019-08-26
中国生物医学工程学报(2019年3期)2019-07-16
大众健康(2017年1期)2017-04-13
中国医科大学学报(2016年11期)2016-12-01
现代电生理学杂志(2016年1期)2016-07-10
磁共振成像(2015年2期)2015-12-23
现代电生理学杂志(2015年3期)2015-07-18
