
国产注射用头孢哌酮钠舒巴坦钠的质量评价
2022-02-19 08:46:34陈蓉马冬阳蔡雪萍苏静华顾晓红郭彬朱乃军许丹郝刚邢以文
中国抗生素杂志 2022年1期
陈蓉 马冬阳 蔡雪萍 苏静华 顾晓红 郭彬 朱乃军 许丹 郝刚 邢以文
(苏州市药品检验检测研究中心,苏州 215104)
头孢哌酮(C25H27N9O8S2)为第三代广谱半合成头孢菌素,通过抑制胞壁黏肽合成酶,从而阻碍细胞壁黏肽合成,使细菌胞壁缺损,菌体膨胀裂解。能对抗多种β-内酰胺酶的降解作用,抗菌谱广,对革兰阳性菌及阴性菌均有作用[1]。舒巴坦钠(C8H10NNaO5S)属于半合成青霉烷砜类衍生物,为不可逆的竞争性β-内酰胺酶抑制剂,是继克拉维酸后第二种用于临床的β-内酰胺酶抑制剂,本身的抗菌活性弱,略强于克拉维酸,单用时仅对淋球菌和不动杆菌属有杀菌作用,但其对多数β-内酰胺类抗生素耐药菌株产生的β-内酰胺酶具有不可逆的抑制作用,因而可保护β-内酰胺类抗生素免受水解破坏[2]。两种药物合用,具有明显的协同作用,其抗菌作用较单用头孢哌酮高4倍[3]。
注射用头孢哌酮钠舒巴坦钠(cefoperazone sodium and sulbactam sodium for injection)是头孢哌酮钠与舒巴坦钠组成的复方制剂。最先由美国辉瑞公司研制,1986年在日本率先上市,1994年9月由大连辉瑞制药有限公司获批原研进口,1997年起国内生产企业相继获批上市。临床上主要用于抗易感菌株引起的中度和重度感染治疗,并限用于有明确药敏试验证据或重症感染的患者。主要治疗由敏感细菌所引起的呼吸道感染、泌尿道感染、腹内感染、败血症、脑膜炎、皮肤及软组织感染、眼部感染、骨骼及关节感染等;预防因腹腔、妇科、心血管、骨科及整形手术所引起的手术后感染[4]。美国胸科学会/感染学会(ATS/IDSA)指南(2019年)推荐亚洲国家将注射用头孢哌酮钠舒巴坦钠作为中重度院内获得性肺炎(HAP)、呼吸机相关性肺炎(VAP)、多重耐药菌株(MDR)、铜绿假单胞菌、肺炎克雷伯菌、不动杆菌属等感染的初始经验用抗生素之一。
注射用头孢哌酮钠舒巴坦钠由头孢哌酮钠舒巴坦钠原料直接无菌分装制成,因此其原料的品质直接影响制剂的质量[5]。中国药典2015年版二部[6]和日本药典JP17版[7]收载了原料及其制剂,欧洲药典(EP)9.0[8]仅收载了原料标准。该品种临床使用非常广泛,连续多年蝉联国内医疗机构抗感染药物使用量TOP1。不良反应发生数较高,常见的严重不良反应为皮疹、瘙痒、呼吸困难、胸闷、凝血障碍、过敏样反应、寒战、过敏反应、血小板减少[9]。随着近年来“限抗令”的实施,对该品种的质量风险控制尤为重要。
目前国内共有注射用头孢哌酮钠舒巴坦钠生产企业89家,批准文号352个,包括11种规格。国家药品监督管理局将本品种列入2020年度国家药品抽验计划。按照要求,我单位对国内39家企业生产的260批次注射用头孢哌酮钠舒巴坦钠进行了法定检验和探索性研究,客观评价该品种的质量现状,查找可能存在的影响品种安全性和有效性的因素,提出质量标准完善建议,为企业提高生产工艺起到积极作用,同时也为上市后的药品监管提供可靠的的技术支撑。
1 仪器与试药
1.1 仪器
Waters Acquity H Class超高效液相色谱仪(美国Waters公司);Waters ACQUITY 2D UPLC液相色谱系统,Xevo G2-XS Q-TOF质谱系统配备MassLynx工作站(美国Waters公司);Agilent 7200 Accurate-Mass Q-TOF GC/MS配备NIST MS Search2.0工作站(美国Agilent公司);Thermo X-Series 2型ICP-MS,配备PlasmaLab工作站(美国Thermo Fisher Scientific公司);HACH TL2350浊度仪(美国Hach公司);Mastersizer 2000智能激光粒度仪(英国Malvern公司);XSE205DU型电子天平(瑞士Mettler Toledo公司);Direct-Q3型超纯水机(美国Millipore公司);SK8210HP型超声波清洗器(上海科导超声仪器有限公司);XMTD-204型数显式电热恒温水浴锅(上海跃进医疗器械有限公司);Metrohm 915KF卡氏水分测定仪(瑞士万通公司)。
1.2 试药
对照品:头孢哌酮、头孢哌酮杂质A、头孢哌酮杂质C(1-甲基-5-巯基四氮唑)、头孢哌酮杂质F(头孢哌酮S异构体)与舒巴坦、N-亚硝基二甲胺,以上均购自中国食品药品检定研究;头孢哌酮杂质B、头孢哌酮杂质D、舒巴坦杂质A、舒巴坦杂质C、舒巴坦杂质E、舒巴坦杂质F为EP对照品; 铅、镉、砷、汞、钴、镍、钒、硒、锂、锑、钡、铬、钼、铜、锡、钯、锌、铁,以上均购自国家有色金属及电子材料分析测试中心;BHT(2,6-二叔丁基-4-甲基苯酚)、八甲基环四硅氧烷均购自Sigma公司。
试剂:乙腈、四丁基氢氧化铵均为色谱纯;冰醋酸、硝酸均为UP级;水为超纯水,其余试剂均为分析纯。
1.3 样品
260批次注射用头孢哌酮钠舒巴坦钠均为2020年国家药品计划抽验样品,涉及39个生产企业。企业覆盖率43.8%、批准文号覆盖率24.1%;抽样覆盖了全国22个省、4个直辖市、5个自治区,抽样数量在各省之间分布均匀。其中从生产企业抽样112批,经营企业抽样139批,医疗机构抽样9批。
2 实验方法
2.1 法定标准检验
此次抽验全部批次按照《中国药典》2015年版二部注射用头孢哌酮钠舒巴坦钠标准检验,部分项目涉及到国家食品药品监督管理局注册标准5个:YBH00672013、YBH11332006、YBH15742005、YBH28592005、WS1-(XG-002)-2005。
2.2 探索性研究
为全面评价注射用头孢哌酮钠舒巴坦钠的质量,完善质控体系,探讨工艺对产品质量的影响,结合企业调研结果、法检结果、以及常见抗生素粉针剂研究方向等参考资料[10-12],围绕药品安全性和有效性两个方面进行相关的探索性研究。
2.2.1 有关物质I
根据EP 9.0头孢哌酮及舒巴坦项下有关物质方法,探索头孢哌酮杂质A、B、C、D、F以及舒巴坦杂质A、C、E、F等杂质的分离,建立统一的UPLC方法进行检查,结合杂质单标对照品定位和破坏试验,考察样品的杂质谱。
色谱条件:色谱柱:Waters Acquity UPLC HSS T3(2.1 mm×100 mm,1.8 μm);以0.05%三氟乙酸水溶液为流动相A,以乙腈为流动相B,按表1梯度洗脱;流速:0.4 mL/min;柱温:35℃;检测波长:230 nm;进样量:2 μL。
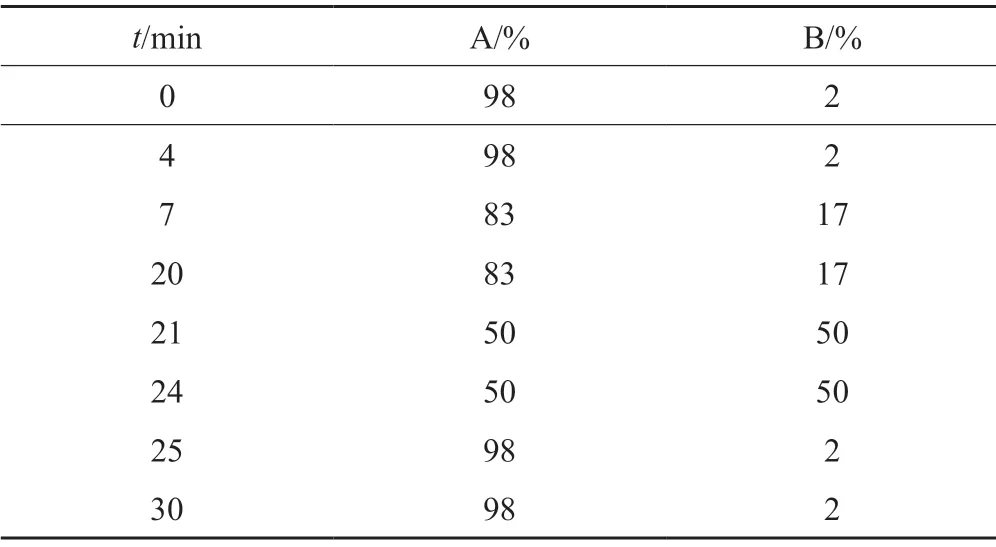
表1 梯度洗脱程序Tab.1 Gradient elution program
2.2.2 有关物质Ⅱ
引发过敏反应的过敏原是β-内酰胺类抗生素中的聚合物类杂质[13]。建立聚合物类杂质的定量分析方法,并采用二维柱切换超高效液相色谱-四级杆/飞行时间质谱(UPLC-Q/TOF-MS)联用技术对其杂质进行结构推断。
第一维色谱条件:色谱柱:TSK-gel G2000SWxl凝胶柱(7.8 mm×300 mm,5 μm);以磷酸盐缓冲液(pH7.0)[0.005 mol/L磷酸氢二钠溶液-0.005 mol/L磷酸二氢钠溶液(61:39)]-乙腈(95:5)为流动相;流速:0.5 mL/min;柱温:25℃;检测波长:254 nm;进样量:20 μL。第二维色谱条件:色谱柱:ACQUITY UPLC HSS T3(2.1 mm×100 mm,1.8μm);以0.005 mol/L甲酸铵水溶液为流动相A,以乙腈为流动相B,按表2梯度洗脱;流速:0.4 mL/min;柱温:25℃;切换用定量环体积:250 μL。
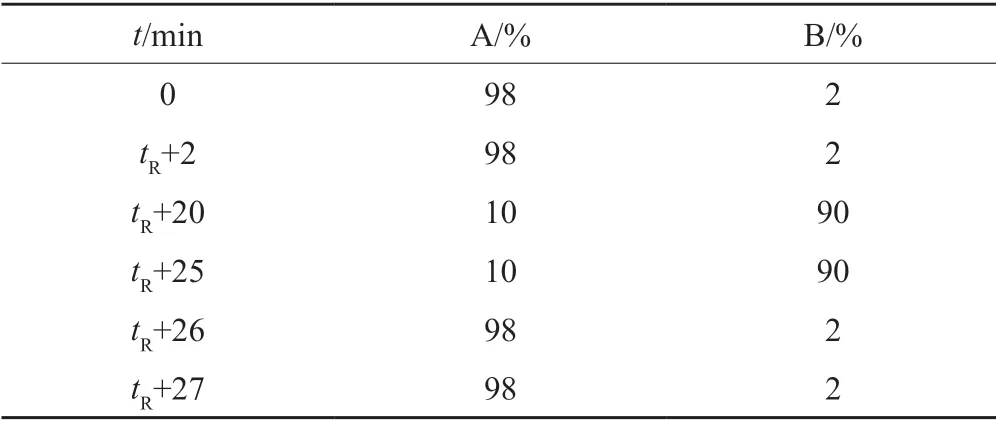
表2 梯度洗脱程序Tab.2 Gradient elution program
质谱条件:ESI离子源;正离子模式;一级质谱扫描范围:m/z50~1500;离子源温度:110℃;毛细管电压:2.50 kV;锥孔电压:40 V;去溶剂气(N2)温度:250℃;流量:600 L/h;锥孔气流:50 L/h。
2.2.3 金属元素的提取和迁移
玻璃由于本身的化学成分及生产工艺等因素,其中的金属离子或阳离子团均有可能从玻璃中迁移出来,造成药品中残留无机金属元素杂质。为了进一步研究样品中金属元素杂质的含量及与工艺等的内在联系,开展了西林瓶及药物中金属元素的提取及迁移量研究。根据元素杂质可能的几种来源,包括合成催化剂、玻璃包材常用元素及ICH相关要求,选择了铅、镉、砷、汞、钴、镍、钒、硒、锂、锑、钡、铬、钼、铜、锡、钯、锌和铁等18种元素作为考察对象,建立ICP-MS法考察39个厂家使用的西林瓶及108批制剂中上述金属元素的提取及迁移量,评价西林瓶的安全性。
2.2.4 胶塞相容性
在药物的贮存过程中,胶塞直接与粉末接触,胶塞中的一些挥发性成分不可避免的与头孢菌素发生相互作用,其与特定挥发性成分的吸附作用是导致注射剂澄清度变差的最主要原因。建立GC-MS法测定各厂家胶塞和制剂中的4种硅氧烷类和2,6-二叔丁基-4-甲基苯酚(BHT)等挥发性成分,采用浊度仪测定溶液的浊度值,同时选取两家企业的头孢哌酮钠原料药、舒巴坦钠原料药进行模拟实验,与胶塞挥发物试剂(BHT和八甲基环四硅氧烷)一同密闭放置1、2和5d后,考察挥发性成分及浊度值变化,探讨两者之间的相关性,为胶塞的选择提供参考。
2.2.5 粒度与粒度分布
药物的粒子大小也是决定其溶解速率的因素之一,采用Malvern Mastersizer 2000激光粒度分析仪对39家企业260批制剂的粒度和粒度分布进行了测定。实验参数:干法测定。背景及样品的扫描时间为10 s;颗粒折射率1.52;颗粒吸收率0.1;分散压力2.0 bar;进样速率50%;遮光度0.5%~5%。
同时考查各厂家制剂复溶时间:取各厂家制剂1支,分别加入注射用水适量,使头孢哌酮含量为125 mg/mL,置震荡器上振摇溶解,转速100 r/min,观察并计时。
2.2.6 影响因素考察
参照中国药典2015年版通则9001原料药与制剂稳定性试验指导原则,考察高温(60℃,30%)、高湿(25℃,90%)、光照(25℃,4000Lx)3种条件下,39家生产企业的53批样品放置1个月后的酸度、有关物质Ⅰ、有关物质Ⅱ等指标的变化情况。
2.2.7 含量均匀度
中国药典2015年版四部通则0941含量均匀度检查法项下规定,药物间或药物与辅料间采用混粉工艺制成的注射用无菌粉末均应检查含量均匀度。虽然注射用头孢哌酮钠舒巴坦钠制剂标准项下并未规定含量均匀度检查,但双组分不同配比的制剂,通过含量均匀度考察可以对药品质量及分装均匀性进行评价,使含量的分析结果更客观。
色谱条件、系统适用性、对照品溶液配制等条件均同法检含量测定。供试品溶液配制:取本品10瓶,分别溶解并定量稀释制成约含头孢哌酮0.5mg/mL溶液。对39个厂家260批不同规格注射用头孢哌酮钠舒巴坦钠进行含量均匀度检查,并对结果进行分析。
3 结果与分析
3.1 法定检验结果
按现行质量标准检验,39家企业的260批次样品均符合规定,合格率100%。本次抽样样品生产时间介于2018年11月至2020年6月之间,由图1可知,检验结果表明该产品质量与生产时间具有一定的相关性,随着样品从生产距离检验时间延长,头孢哌酮杂质A、头孢哌酮杂质C、有关物质Ⅱ杂质呈变大的趋势,溶液的澄清度从浅于0.5号浊度标准液逐渐加深至浅于1号浊度标准液,溶液的颜色由浅于黄色或黄绿色2号标准比色液逐渐加深至浅于黄色或黄绿色4号标准比色液。本次从不同抽样点抽取同企业同批号样品12组(24批),对其酸度、水分、头孢哌酮杂质A、杂质C、其他最大单杂、其他总杂、头孢哌酮含量、舒巴坦含量8个检验指标结果进行了SPSS配对t检验,结果表明同批号样品检验结果间无显著差异(P>0.05),表明在规定的储藏条件下(密闭,在凉暗干燥处)保存,样品质量与抽样地无明显相关性,具体结果见表3。
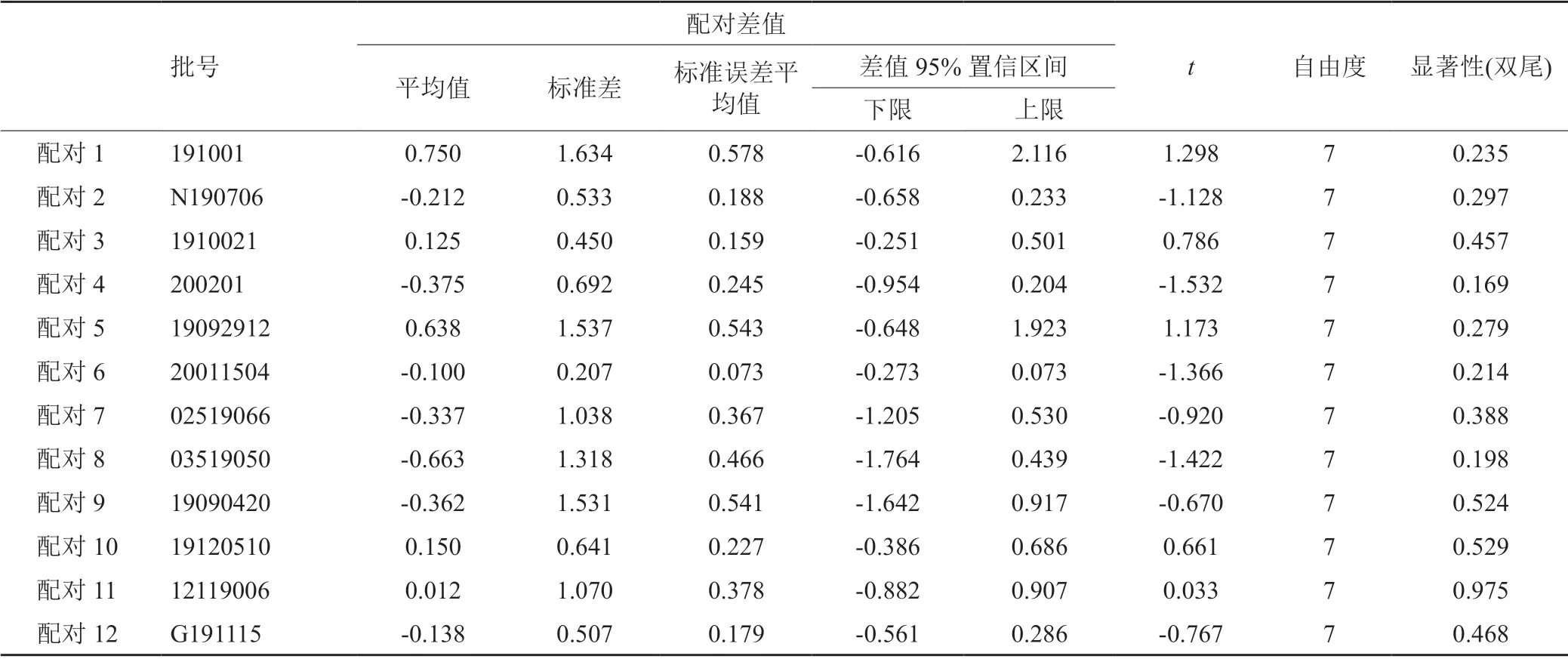
表3 同批号配对样本检验Tab.3 Paired sample test of same batch samples
3.2 探索性研究结果
3.2.1 有关物质I
《中国药典》2015年版二部仅控制头孢哌酮杂质A、头孢哌酮杂质C两个已知单杂及其他单杂、其他总杂,典型供试品色谱图如图2。采用“2.2.1”项下建立的杂质谱分析方法共检出9个杂质峰,各杂质峰与2个主峰(峰3为舒巴坦峰,峰9为头孢哌酮峰)分离度较好,如图3~4,溶剂空白表明基线基本无干扰,9种成分在各自标曲范围内均呈现良好线性(r≥0.9998),精密度、重复性、回收率均满足要求(RSD<2.0%),头孢哌酮杂质A、B、C、D、F和舒巴坦杂质A、C、E、F检出限分别为:0.022、0.313、0.093、0.316、0.319、0.029、0.836、2.194和0.265 μg/mL。经检测260批样品:有27批检出了舒巴坦杂质A(峰1),按舒巴坦标示量计为0.07%~0.18%;有108批检出了头孢哌酮杂质F(峰10),按头孢哌酮标示量计为0.05%~0.22%;头孢哌酮杂质A(峰7),按头孢哌酮标示量计为0.3%~1.0%。头孢哌酮杂质C(峰2),按头孢哌酮标示量计为0.1%~0.5%;其他最大单杂按自身对照法计为0.1%~0.5%;其他杂质总和按自身对照法计为0.4%~2.4%。
由图5可知,与现行标准有关物质检测方法相比,新建方法的头孢哌酮杂质A和头孢哌酮杂质C结果差异不明显;其他最大单杂、其他杂质总和的检出数量和含量均明显增加,且检出的杂质含量平均偏差小于法检方法。由于杂质多样性以及检测手段的局限性,UPLC法仍需要进一步进行研究论证,方可作为该品种有关物质检查标准提高的补充。
3.2.2 有关物质Ⅱ
按“2.2.2”项下方法检验,测定结果在0.81%~2.11%之间。由图6可知,在TSK柱的测定条件下,共检出4种杂质,经LC-MS分析,杂质1和杂质2未检出明显的杂质峰,未做定性推测;杂质3检出1个杂质峰,杂质4共检出4个杂质峰,各杂质可能的化学结构及来源分析:杂质3可能为头孢哌酮与其降解杂质及工艺杂质形成的三聚物,杂质4a可能为头孢哌酮降解物或衍生物与头孢哌酮杂质E的聚合物,杂质4b、4c、4d为同分异构体,可能为头孢哌酮与其降解杂质及工艺杂质二聚物,具体杂质推测见表4。
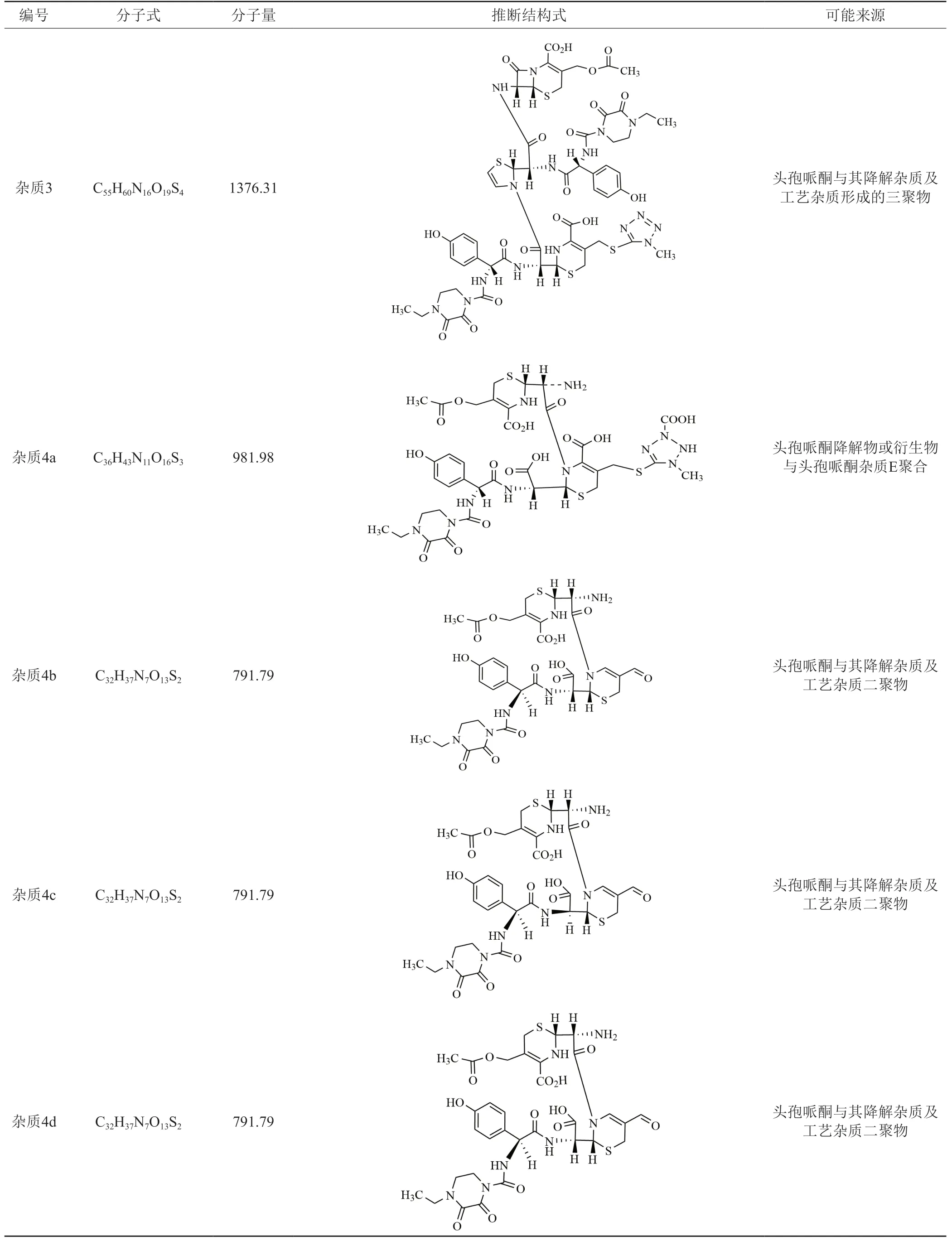
表4 各杂质可能的化学结构及来源分析Fig.4 Possible chemical structures and sources of impurities
现行标准仅YBH00672013对其聚合物类杂质(有关物质II)进行了控制。由上述方法测定发现,主峰前的色谱峰并不都是聚合物杂质,个别色谱峰也不是纯峰,而是多个化合物(杂质)的混合物[14]。有关物质Ⅰ不能全面反映该品种的全部杂质情况。有关物质Ⅱ既能检查聚合物类杂质,又可作为有关物质Ⅰ检查的一个补充,可完善杂质谱,以减少不良反应的发生。
3.2.3 金属元素的提取和迁移
39个厂家西林瓶离子检测结果可以得出,除钴、钼、汞、钯、硒5种离子均无检出,其余13种离子部分厂家有检出。其中低硼硅玻璃管制注射剂瓶的Ba、Zn、Fe含量显著高于钠钙玻璃模制注射剂瓶。钠钙玻璃模制注射剂瓶浸出的金属离子数量和含量均较低硼硅玻璃管制注射剂瓶偏低,理论上来说相对更安全。与《化学药品注射剂与药用玻璃包装容器相容性研究技术指导原则(试行)》及ICH Q3D “元素杂质指导原则”规定的PDE(每日允许暴露量)相比,39家企业采用的西林瓶浸出离子含量均符合规定。108批制剂离子检测结果显示,除锑、钯、硒3种离子均无检出,其余15种离子大部分厂家均有检出,但离子浓度均无明显的变化趋势,说明药液与注射剂瓶内表面之间未发生明显迁移。可认为浸出物的量不会改变药品的安全性,对患者的安全性风险小。表明目前采用的各类西林瓶内包材与该品种制剂相容性较好。
3.2.4 胶塞相容性
由图7可知,浊度较大的样品,其挥发物中含有的硅氧烷类和BHT越多,表明制剂溶液的浊度值与硅氧烷类和BHT具有正相关性,提示生产企业在选择胶塞时,应着重考察胶塞中硅氧烷类和BHT的含量。模拟吸附试验表明,头孢哌酮钠吸附的BHT含量与浊度值有一定的相关性,舒巴坦钠吸附的硅氧烷类和BHT含量与浊度值几乎没有相关性,即硅氧烷类和BHT对溶液澄清度的影响主要作用于头孢哌酮钠。结合法检和探索性研究得出,影响溶液澄清度的因素主要为药品与胶塞作用的时间、规格、胶塞种类等。头孢哌酮钠与胶塞作用时间延长,浊度值增大;小规格制剂与胶塞接触更为充分,随着时间的推移,胶塞中的硅氧烷类和BHT迁移到药物粉末中的更多,浊度值越大;在胶塞选择时尽可能选择覆膜胶塞。国内两家企业的0.5g规格制剂,采用浊度仪法测定溶液的澄清度,均超过了1号浊度标准液,由于同一厂家不同规格制剂均使用相同胶塞,药物粉末与胶塞接触而发生的相互影响是一样的,但在澄清度时加水量不一样,规格小的制剂可能澄清度较其他规格的差,存在影响药品安全性的质量风险,应密切注意该风险。
3.2.5 粒度与粒度分布
由图8复溶时间研究表明,原研样品(9号)在60秒内全部溶解,大部分企业产品在60~120 s内溶解,小部分厂家制剂120s后仍有残留未溶解。目前该品种国内主要有3大原料药生产厂家,分别为:SZDR、QLAT、ZHLB,通过比对各厂家原料药来源发现,编号为1、3、4、6、10、12、17、15、18、19、25、27、28、29、30、31、33的制剂原料药来自于SZDR,普遍溶解较快;编号为2、8、22、23、26、34的制剂原料药来自于ZHLB,溶解时间最长;其余来自QLAT和其他原料药厂家,溶解时间多在1~2 min内。推测不同厂家制剂复溶行为存在差异,可能与其原料药不同有关。
各厂家制剂的粒度分布范围为D(0.1)=0.8~4.3 μm,D(0.5)=8.7~63.6 μm,D(0.9)=122.1~581.6 μm,原研药品的90%粒径范围为197.8~361.9 μm,最大粒径373.6 μm,来自SZDR原料药的制剂企业的90%粒径范围为122.1~336.4 μm,最大粒径380.4 μm,来自QLAT原料药的制剂企业的90%粒径范围为169.4~412.4 μm,最大粒径441.7 μm,来自ZHLB原料药的制剂企业的90%粒径范围为236.4~581.6 μm,最大粒径612.6 μm。原研药品的粒径比国产药品的更小且分布较为均匀集中,推测可能更利于药品的溶解,结果见表5。D(0.5)和D(0.9)的分布与头孢哌酮和舒巴坦的配比具有相关性。如图9所示,同一厂家,头孢哌酮与舒巴坦配比为2:1规格的粒径通常大于1:1规格。粒度分布考察可为处方配比及制剂工艺优选提供依据,提示企业在生产过程中进一步加强对原料和生产工艺的控制。
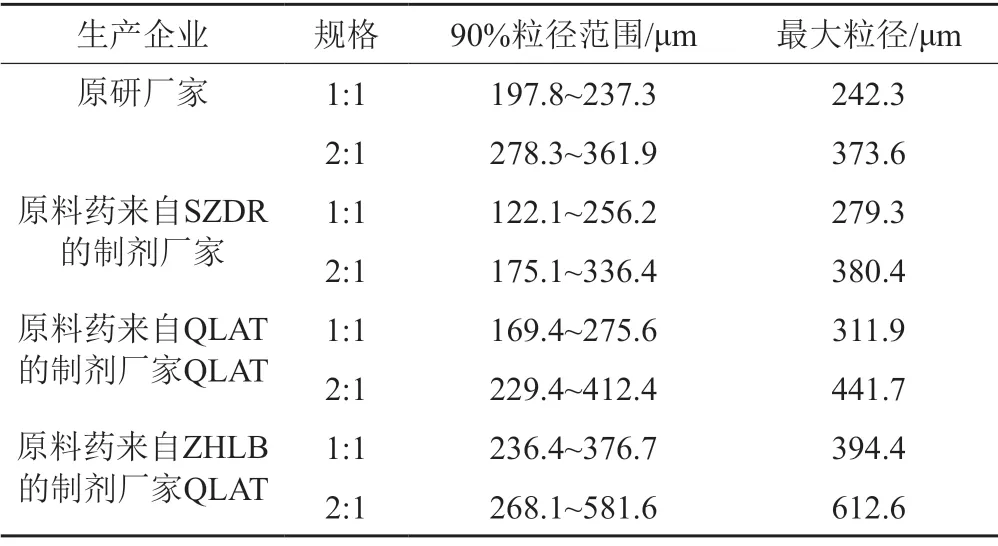
表5 粒径分布分析结果Tab.5 Analysis of the particle size distribution of the samples
3.2.6 影响因素考察
高温、高湿、光照处理后的样品pH值均下降,其中高温影响最显著,高湿和光照结果相当;头孢哌酮杂质A、头孢哌酮杂质C、其他最大单杂、其他总杂、头孢哌酮杂质F、舒巴坦杂质A 6个指标受高温影响显著增加;除舒巴坦杂质A对高湿和光照敏感外,其余5个指标在经过高湿或光照处理后,变化趋势不明显;高温处理后的样品有关物质Ⅱ含量显著增长,高湿处理后基本没变化,光照处理后略有增加。综上所述,温度是影响pH、杂质的最关键因素,提示生产、经营、使用各环节在储存过程中必须注意温度监控。
3.2.7 含量均匀度
259批制剂两种主成分的含量均匀度结果良好,A+2.2S均小于15.0;一厂家1批规格2.25g(2:1)制剂的舒巴坦含量均匀度A+2.2S超标,可能是混粉分装工艺存在问题,同时发现该厂家药品的粒径分布也相对较大,头孢哌酮与舒巴坦颗粒大小的悬殊也容易导致混粉不均匀。如图10,头孢哌酮和舒巴坦含量总体呈正态分布,其中头孢哌酮分布的中心点偏向于100%的左侧,而舒巴坦分布的中心点则偏向于100%的右侧。推测可能是生产时头孢哌酮投料量可能未达到100%,而舒巴坦则可能超过100%;抑或是头孢哌酮更容易降解,导致测得的含量低于100%。含量均匀度考察能更全面客观地反映产品含量的真实情况,尤其适用于双组分混合比例差距较大的粉针剂品种,因此建议在注射用头孢哌酮钠舒巴坦钠质量标准中增加含量均匀度检查。
4 结论
目前,注射用头孢哌酮钠舒巴坦钠的法定质量标准基本合理,限度较为合适,能够较好的控制本品的质量。按现行标准检验,本次抽验的39家生产企业260批样品,均符合标准规定。探索性研究修订了有关物质检查、增加了含量均匀度和有关物质Ⅱ等项目。采用拟修订的标准对260批样品检验,合格率99.6%。产品总体质量较好。
注射用头孢哌酮钠舒巴坦钠为我国抗感染类药物临床使用量排名第一的品种,不良反应也相对多发。本品生产厂家繁多,但制剂工艺相对简单统一,此次抽验涉及中国药典2015年版二部及5个企业注册标准,上述标准主要区别在于部分企业在执行中国药典2015年版二部的基础上,按各自企业注册标准执行异常毒性项目,且部分项目执行的限度不一致。各标准之间无较大差异,不利于整体质量控制,均未能有效地控制有关物质和聚合物杂质,缺乏含量均匀度等检查。探索性研发现,产品在生产工艺、内包材、说明书等方面存在缺陷,临床用药有安全隐患。因此建议企业有条件时:①对法定标准进行规范统一;②修订有关物质检查方法和控制限度;③增订包含聚合物的有关物质Ⅱ检查项;④对其产品的分装工艺进行研究,提高批间稳定性;⑤针对小规格制剂胶塞进一步开展包材相容性研究;⑥进行原料药晶型和基因毒性杂质研究;⑦尽快开展一致性评价工作。
此外,该品种目前共有批准文号352个,本次抽样仅涉及不足三成的文号,未抽到的大多数批准文号可能已经闲置,造成的不仅是社会资源的浪费,而且企业长期不生产,偶尔生产可能会存在质量风险,对药品生产监管、上市药品再评价、不良反应监测非常不利。因此建议企业梳理批准文号,对于长期不生产的品种应当查找原因;建议药监部门对此类情况进行排查,加强监管,保障药品质量。
猜你喜欢
中国药学药品知识仓库(2022年7期)2022-05-10 11:03:13
昆明医科大学学报(2021年6期)2021-07-31 07:40:34
天然气工业(2018年11期)2018-12-03 01:15:12
国外医药(抗生素分册)(2016年5期)2016-07-12 14:25:32
国外医药(抗生素分册)(2016年1期)2016-07-10 12:02:35
实用中西医结合临床(2015年7期)2015-02-28 16:30:39
机电信息(2014年26期)2014-02-27 15:53:37
石油矿场机械(2010年1期)2010-12-11 02:47:48
