
Observational Study ldentification of functional tumor necrosis factor-alpha promoter variants associated with Helicobacter pylori infection in the Sudanese population:Computational approach
2022-02-15 07:52AbeerBabikerIdrisAlaaIdrisManalGumaaMohammedBabikerIdrisAmandaElgoraishMohamedMansourDaliaAllamBashirMOArbabNazarBeiragEIAminIbrahimMohamedHassan
Abeer Babiker Idris,Alaa B Idris,Manal A Gumaa,Mohammed Babiker Idris,Amanda Elgoraish,Mohamed Mansour,Dalia Allam,Bashir MO Arbab,Nazar Beirag,EI-Amin M Ibrahim,Mohamed A Hassan
Abeer Babiker ldris,Department of Agricultural Science and Technology,Institute of Natural and Applied Sciences,Erciyes University,Kayseri 38039,Turkey
Abeer Babiker ldris,Department of Medical Microbiology,Faculty of Medical Laboratory Sciences,University of Khartoum,Khartoum 11111,Sudan.abeer.babiker89@gmail.com
Alaa B ldris,Department of Neurosurgery,Ribat University Hospital,Khartoum 11111,Sudan
Manal A Gumaa,El-Amin M lbrahim,Department of Medical Microbiology,Faculty of Medical Laboratory Sciences,University of Khartoum,Khartoum 11111,Sudan
Mohammed Babiker ldris,BioMérieux Clinical and Application Advisor,Al-Jeel Medical Co.,Riyadh 11422,Saudi Arabia
Amanda Elgoraish,Department of Epidemiology,Tropical Medicine Research Institute,Khartoum 11111,Sudan
Mohamed Mansour,Dalia Allam,Department of Gastroenterology,Ibn Sina Specialized Hospital,Khartoum 11111,Sudan
Bashir MO Arbab,Department of Gastroenterology,Modern Medical Centre,Khartoum 11111,Sudan
Nazar Beirag,Biosciences,College of Health,Medicine and Life Sciences,Brunel University,London UB8 3PH,Uxbridge,United Kingdom
Mohamed A Hassan,Department of Bioinformatics,Africa city of technology,Khartoum 11111,Sudan
Mohamed A Hassan,Department of Bioinformatics,DETAGEN Genetic Diagnostics Center,Kayseri 38350,Turkey
Mohamed A Hassan,Department of Translation Bioinformatics,Detavax Biotech,Kayseri 38350,Turkey
Abstract BACKGROUND Helicobacter pylori (H.pylori) is a ubiquitous bacterium that affects nearly half of the world’s population with a high morbidity and mortality rate.Polymorphisms within the tumor necrosis factor-alpha (TNF-A) promoter region are considered a possible genetic basis for this disease.AIM To functionally characterize the genetic variations in the TNF-A 5’-region (-584 to+107) of Sudanese patients infected with H.pylori using in silico tools.METHODS An observational study was carried out in major public and private hospitals in Khartoum state.A total of 122 gastric biopsies were taken from patients who had been referred for endoscopy.Genomic DNA was extracted.Genotyping of the TNF-A-1030 polymorphism was performed using PCR with confronting two-pair primer to investigate its association with the susceptibility to H.pylori infection in the Sudanese population.Furthermore,Sanger sequencing was applied to detect single nucleotide polymorphisms in the 5’-region (-584 to +107) of TNF-A in H.pylori-infected patients.Bioinformatics analyses were used to predict whether these mutations would alter transcription factor binding sites or composite regulatory elements in this region.A comparative profiling analysis was conducted in 11 species using the ECR browser and multiple-sequence local alignment and visualization search engine to investigate the possible conservation.Also,a multivariate logistic regression model was constructed to estimate odds ratios and their 95% confidence intervals for the association between TNF-A-1030,sociodemographic characteristics and H.pylori infection.Differences were statistically significant if P < 0.05.Statistical analyses were performed using Stata version 11 software.RESULTS A total of seven single nucleotide polymorphisms were observed in the TNF-A 5’-region of Sudanese patients infected with H.pylori.Only one of them (T > A,-76)was located at the in silico-predicted promoter region (-146 to +10),and it was predicted to alter transcription factor binding sites and composite regulatory elements.A novel mutation (A > T,+27) was detected in the 5’ untranslated region,and it could affect the post-transcriptional regulatory pathways.Genotyping of TNF-A-1030 showed a lack of significant association between -1030T and susceptibility to H.pylori and gastric cancer in the studied population (P=0.1756) and (P=0.8116),respectively.However,a significant association was detected between T/C genotype and H.pylori infection (39.34% vs 19.67%,odds ratio=2.69,95% confidence interval:1.17-6.17,P=0.020).Mammalian conservation was observed for the (-146 to +10) region in chimpanzee (99.4%),rhesus monkey (95.6%),cow (91.8%),domesticated dog (89.3%),mouse (84.3%),rat(82.4%) and opossum (78%).CONCLUSION Computational analysis was a valuable method for understanding TNF-A gene expression patterns and guiding further in vitro and in vivo experimental validation.
Key Words:5’-region;Promoter;TNF-A;Helicobacter pylori;In silico analysis;Sudan
lNTRODUCTlON
Helicobacter pylori(H.pylori) is a widespread bacterium that affects nearly half of the world’s population with a high morbidity and mortality rate[1].Generally,the decreasing prevalence ofH.pyloriinfection is continuing in many parts of North America and Europe.Still,no such decline has been noted in most developing countries,including Sudan,in which theH.pyloriinfection represents a critical public health challenge[2-4].Although there is a common consensus that the risk of acquisition and transmission ofH.pylorican be minimized and prevented to a large extent,this drop in those countries,whether it is a local phenomenon or a national trend,has to be investigated[2].
Infection withH.pyloriis usually acquired during childhood.However,it may persist if not treated in the host’s stomach throughout life due to the synchronized balance between the bacteria and its host[5,6].The long duration ofH.pyloriin the gastric environment will develop chronic gastritis inH.pylori-infected persons.At the same time,a few may be complicated to more severe diseases such as peptic ulcer,atrophic gastritis,mucosa-associated lymphoid tissue lymphoma or gastric adenocarcinoma[7,8].The variability inH.pyloriinfection outcomes is determined by bacterial factors,genetic characteristics of the host,especially those regarding polymorphisms in specific cytokines,and the environmental factors[7,9,10].However,the crucial role of cytokine-related polymorphisms in the susceptibility or severity ofH.pyloriinfection has been attracting considerable attention after El-Omaret al[11] proposed that functional polymorphisms in proinflammatory cytokine genes inH.pyloriinfected patients are associated with a two-to three-fold increased gastric cancer risk.Because tumor necrosis factor-alpha (TNF-a) production has been implicated as an essential factor in immune regulations and inflammatory responses againstH.pyloriinfection[12],polymorphisms within theTNF-Apromoter are considered a possible genetic basis for this disease[13].
TNF-a is a potent pleiotropic proinflammatory cytokine that mediates diverse biological and pathological processes,in addition to its responsibility for the regulation of two opposite processes:proliferation and apoptosis (reviewed in[14,15]).TNFgene transcription is regulated by nucleoprotein complexes known as enhanceosomes that involve distinct sets of transcription factors and coactivators at the proximalTNFpromoter and in the pattern of chromosomal organization of theTNF/lymphotoxinlocus that function in synergy to drive transcription[16].TNFenhanceosome assembly is cell type-and stimulus-specific,partially because of differential occupancy of overlapping DNA motifs in theTNFpromoter.For example,after lipopolysaccharide stimulation of monocytes where no inducible translocation of NFATp can be detected,Sp1 and Ets/Elk proteins can successfully bind to sites that NFATp occupies in T cells[13,14].Furthermore,manyTNFactivator binding motifs can be recognized by more than one class of transcription factor depending on the ambient concentration of different elements in the nucleus and the stimulus and cell type[16].These features provide theTNFpromoter with a remarkable degree of flexibility to respond to multiple stimuli in a specific manner through a short cisregulatory region[13].
TheTNFlocus lies within the major histocompatibility complex region on the short arm of human chromosome 6p21.3,a highly polymorphic region[17].Many bi-allelic polymorphisms and microsatellites are found at or around theTNFlocus[14,18].Several have been suggested to be implicated in diseases susceptibility/resistance or severity by modifying the transcriptional regulation ofTNF[13,18].There are different transition variants in theTNF-Apromoter region at the positions of -237,-307,-375,-856,-862 and -1030 in which the positions -307 and -237 have been most frequently evaluated for the association withH.pyloriinfection (susceptibility or progression)[13].It has been reported that the presence of the adenine allele at -237 or -307 positions causes higher constitutive and inducible transcriptional levels than the guanine allele,which results in enhanced production of TNF-a up to five-foldin vitro[19,20].In addition,the single nucleotide polymorphism (SNP) at the -1030 position,which is frequently observed in African populations[21,22],has been suggested to be related to high TNF-α production[23].However,the influence of these SNPs on TNF-α production is not fully known and still a contradictory topic of debate,which is explained,in part,by ethnic differences and genetic variations between populations[19,20].
To our knowledge,there are no previous studies in Sudan that have addressed the association betweenTNF-AandH.pyloriinfection orH.pylori-associated diseases.Therefore,in this study,we applied DNA Sanger sequencing to detect genetic variations in theTNF-A5’-region (-584 to +107) of Sudanese patients infected withH.pyloriand predicted if these SNPs could alter transcription factor binding sites (TFBSs)or composite regulatory elements (CEs) using a computational approach.In addition,a comparative profiling analysis was performed to investigate the conservation of these SNPs in 11 species.Regulation mechanisms of TNF-α production may be linked to the difference in responses toH.pyloriinfection.This emphasizes the importance of studying the regulation ofTNF-Agene expression.
MATERlALS AND METHODS
Research methodology
In the present study,in silico analysis was employed for theTNF-A5’-region (-584 to+107) of the Sudanese patients infected withH.pyloriin two steps:(1) In silico prediction of the promoter region;and (2) In silico analysis of the predicted promoter region (-146 to +10).To obtain accurate results,we applied various software/servers to predict the promoter region,CpG island and regulatory motifs because promoters have complex and specific architecture and contain multiple transcription factors involved in particular transcription regulation[24].Therefore,the unique composition of TFBSs and different features of each promoter may lead to different powers and algorithms for promoter identification[25].In addition,genotyping of theTNF-A-1030 T>C polymorphism was performed to assess its association withH.pyloriinfection in the Sudanese population using PCR with confronting two-pair primer method.
However,the initial studies have numbered the positions of SNPs relative to theTNFmRNA cap site incorrectly (e.g.,-308 instead of -307 and -238 instead of -237)[13].In contrast,the corrected numbering should have proceeded 5’ from the adenine at the+1 position[26] as in the Cytokine Gene Polymorphism In The Human Disease Database[27],and we used it throughout this study.The methodology followed in this study is represented in Figure 1.

Figure 1 Schematic representation of the study methodology.H.pylori:Helicobacter pylori;-ve:Negative;+ve:Positive;CTPP:Confronting two-pair primer;SNP:Single nucleotide polymorphism;TNF-A:Tumor necrosis factor-alpha;TFBS:Transcription factor binding site;CE:Composite regulatory elements.
Study population and study setting
An observational study was carried out in major public and private hospitals in Khartoum state in 2019-2020.These hospitals included Soba teaching hospital,Ibin Sina specialized hospital,Modern Medical Centre,Al Faisal Specialized Hospital and Police hospital.In addition,the molecular laboratory processes were conducted in the Molecular Biological Research laboratory at the Faculty of Medical Laboratory Sciences at the University of Khartoum.
The matched case-control formula was selected to calculate the sample size using Epi Info software version 7[28,29],assuming 95% confidence interval (CI),80% power of the study,one ratio of control to the case,15% of controls exposed,3.36 odds ratio(OR) and 37.2% of cases exposed.According to the sample size calculation,the present study included 122 patients referred for upper gastrointestinal tract endoscopy,and most of them were because of dyspepsia.Out of that,61 wereH.pylori-negative patients and regarded as controls.The demographic characteristics of the study population are shown in Table 1.
The diagnosis of gastroduodenal diseases was based on the assessment of an experienced gastroenterologist,and in cancerous cases,histological examinations were also performed to confirm the diagnosis.The gastric pathologies of the study population are presented in Table 2.

Table 1 Demographic characteristics of the study population,n (%)

Table 2 Gastric pathologies of the study population,n (%)
The study population’s demographic and clinical data were obtained by personal interviews,a structured questionnaire and a review of case records.The selection criteria included the Sudanese population from both sexes who were not taking antibiotics or nonsteroidal anti-inflammatory drugs.Written informed consent was granted by the participants after they were informed of the objectives and purposes of the study.
DNA extraction and PCR amplification of specific 16S rRNA gene of H.pylori
For DNA extraction purposes,gastric biopsies were collected in 400 μL of phosphate buffered saline.For histological investigation,the biopsies were preserved in 10%formalin.DNA extraction was performed by using innuPREP DNA Mini Kit(analytikjena AG,Germany) and followed the protocol that was given by the manufacturer,as previously described in[30].
Extracted DNA was amplified for the specific16SrRNA gene to diagnose and confirm the infection ofH.pyloriusing the following primers (primers:F:5’-GCGCAATCAGCGTCAGGTAATG-3’;and R:5’-GCTAAGAGAGCAGCCTATGTCC-3’)[31] and the previously described PCR condition[4].The amplified product for the16S rRNAis 522 bp.
Conventional PCR amplification and sequencing of the TNF-A promoter region
To amplify the promotor polymorphisms -308 and -238 of theTNF-Agene,primers:F:5’-GCTTGTCCCTGCTACCCGC-3’ and R:5’-GTCAGGGGATGTGGCGTCT-3’ were used[32].The following thermal cycling conditions were used:initial denaturation at 95 °C for 5 min,followed by 35 cycles of 94 °C for 60 s,58 °C for 30 s and 72 °C for 60 s,with a final extension at 72 °C for 10 min.The amplified PCR product is 691 bp located between -585 bp upstream and 107 bp downstream of theTNF-Agene.
Fourteen PCR products ofH.pylori-infected patients,which had the clearest bands,were sent for commercial DNA purification and Sanger dideoxy sequencing by Macrogen Inc.,Korea.
Sequence analysis and SNPs detection
Two chromatograms (forward and reverse) for each sequence were visualized,checked for their quality and analyzed with the Finch TV program version 1.4.0[33].In addition,the Basic Local Alignment Search Tool nucleotide (https://blast.ncbi.nlm.nih.gov/)was used to look for and assess nucleotide sequence similarities[34].
For SNP detection in theTNF-Apromoter region,multiple alignment sequences were performed for the tested sequences with a reference sequence (TNF-A;NG_007462) using Clustal W[35].
Bioinformatics analysis of the TNF-A promoter region in H.pylori-infected patients
In silico prediction of the promoter:In this study,we employed five programs for human promoter prediction and recognition of human PolII promoter region and start of transcription (TSS) which include:Berkeley Drosophila Genome Project (http://www.fruitfly.org/)[36],Promoter 2.0 Prediction Server (http://www.cbs.dtu.dk/)[37],FPROM,TSSG and TSSW (http://softberry.com/) which are based on neutral network and linear discriminant approach[25,38,39].
In silico analysis of the predicted promoter region:Assessment for the presence of promoter-associated features:The computationally predicted promoter regions were additionally assessed for the presence of promoter-associated features using the ENCODE data (https://www.encodeproject.org/)[40-42].These features include promoter-associated histone marks,DNase I hypersensitivity clusters,broad chromatin state segmentation,transcription factor ChIP-seq and CpG islands.
Prediction of CpG islands:A CpG island is a DNA segment with high GC and CpG dinucleotide content and often regarded as a marker for the initiation of gene expression.In this study,we applied MethPrimer and GpC finder software (http://w ww.softberry.com/berry.phtml?topic=cpgfinder&group=programs&subgroup=prom oter) to predict CpG islands using default search parameter values for the software:CpG island length >200 bp,CG% > 50%,and Obs/Exp > 0.6[43,44].
Prediction of TFBSs:Prediction of the potentially functional TFBSs is a crucial step in the chain of promoter analytical events.Therefore,we applied five prediction software programs to analyze the predicted promoter region for possible TFBSs:(1)AliBaBa2 (http://www.gene-regulation.com/)[45];(2) Alggen Promo (http://alggen.lsi.upc.es/cgi-bin/promo_v3/)[46,47];(3) TF-Bind (http://tfbind.hgc.jp/)[48];(4)Tfsitescan (http://www.ifti.org)[49];and (5) Gene Promoter Miner (GPMiner) (http://GPMiner.mbc.nctu.edu.tw/),which is an integrated system.For predicting TFBSs,MATCH tool was utilized to scan TFBSs in an input sequence[50].
Prediction of CEs:CE is the minimal functional unit composed of two closely located DNA binding sites for distinct transcription factors,but its regulatory function is qualitatively different from the regulation effects of either individual DNA binding site.Thus,the MatrixCatch algorithm (http://gnaweb.helmholtz-hzi.de/cgibin/MCatch/MatrixCatch.pl) was used to identify the CEs in our region (-146 to +10)[51].
Comparative profiling analysis:ECR Browser (http://ecrbrowser.dcode.org)[52],NCBI Basic Local Alignment Search Tool nucleotide (http://blast.ncbi.nlm.nih.gov/Blast.cgi) and Clustal W (https://www.genome.jp/tools-bin/clustalw)[35] were utilized to analyze the possible conservation of the predicted promoter region.The conservation was assessed in 11 species which include:Cow (Bos taurus),chimpanzee (Pan troglodytes),rhesus monkey (Macaca mulatta),dog (Canis lupus familiaris),mouse (Mus musculus),rat (Rattus norvegicus),chicken (Gallus gallus),zebrafish (Danio rerio),frog (Xenopus laevis),opossum (Monodelphis domestica),fugu pufferfish (Takifugu rubripes) and spotted green pufferfish (Tetraodon nigrovoridis).Furthermore,multiplesequence local alignment and visualization search engine (https://mulan.dcode.org/)was used to evaluate the conservation of the promoter’s SNPs and to screen the possible conservation of TFBSs at these SNP locations[53].
Molecular detection of the TNF-A-1030 C/T polymorphism using PCR with confronting two-pair primer method
Two sets of primers were used in PCR with confronting two-pair primer to detect theTNF-A-1030 C/T polymorphism as follow:for allele T,the sequences of the primers were F:5'-AAGGCTCTGAAAGCCAGCTG-3' and R:5'-CCAGACCCTGACTTTTCCTTCA-3';and for allele C,F:5'-GAAGCAAAGGAGAAGCTGAGAAGAC-3' and R:5'-CTTCCATAGCCCTGGACATTCT-3'[54].The PCR mixture components were the same as in[30].The amplification conditions were initial denaturation at 95 °C for 10 min,followed by 30 cycles of denaturation at 95 °C for 1 min,annealing at 66 °C for 1 min,extension at 72 °C for 1 min and final extension at 72°C for 5 min.The resulting PCR products were visualized by electrophoresis on a 1%agarose gel containing ethidium bromide.The predicted size was confirmed using a 50 bp DNA molecular weight marker.Genotyping was distinguished as follows;444 bp and 316 bp for TT genotype,444 bp,316 bp and 174 bp for TC genotype and 444 bp and 174 bp for CC genotype.
Statistical analysis
Fisher’s test orχ2test investigated Hardy-Weinberg equilibrium of allele and genotype distributions.Descriptive statistical analysis was applied to calculate percentages,proportions and means ± standard deviation.Regarding the incidence ofH.pyloriinfection,differences in frequency distribution by age were examined by the Mann-Whitney test,while differences in the frequency distribution of the categorical demographic and clinical variables of the study population were assessed by Fisher’s test orχ2test.A multivariate logistic regression model was constructed to estimate OR and their 95%CIs for the association betweenTNF-A-1030,sociodemographic characteristics andH.pyloriinfection.Differences were considered to be statistically significant ifP< 0.05.The statistical analyses were performed using Stata version11(StataCorp,College Station,Texas) software.
RESULTS
Nucleotide variations in the 5’-regulatory region (-585 to +107) of the TNF-A gene
In the SudaneseH.pylori-infected patients,a total of seven nucleotide variations were detected in the 5’-regulatory region (six substitution mutations and one insertion mutation of C at +70).Among which,five were bimodally mutated heterozygous SNPs,and two are homozygous SNPs,as illustrated in Table 3 and Figure 2.The results of the multiple sequence alignment were visualized in Jalview[55].The nucleotide sequences of theTNF-A5′-region (-584 to +107) were deposited in the GenBank database under the following accession numbers:from MW251712 to MW251725.
In silico prediction of the TNF-A promoter regions
Five types of promoter prediction programs were applied to predict the promoter regions of theTNF5’-region (-584 to +107),and the results are overviewed in Table 4.Neural Network Promoter Prediction (NNPP version 2.2) predicted one promoter region,located at +1 bp relative to the TNF-α mRNA cap site[26].In addition,FPROM and TSSG programs predicted two promoter regions -146 bp and +5 bp,respectively.The TSSW program also expected these two regions.In comparison,no promoter region was predicted by Promoter 2.0 Prediction Server.
In silico analysis of predicted TNF-A promoter region
Presence of promoter-associated features:ENCODE data revealed a high level of transcription factor occupancy patterns,promoter-associated histone modifications and DNase I hypersensitivity at the in silico predicted promoter region,from -146 to+10 nt relative to the TNF-α mRNA cap site[26].Furthermore,CpGFinder and MethPrimer software were employed to predict the presence of CpG islands.However,both software predicted no CpG islands present in the predicted promoter region,and ENCODE data confirmed no presence of the CpG island in the predicted promoter (see Figure 3).
Conservancy of the predicted promoter:As illustrated in Figure 4,the ECR Browser showed mammalian conservation for the (-146 to +10) region in chimpanzee (Pan troglodytes-pan-Tro2) (99.4%),rhesus monkey (Macaca mulatta-rheMac2) (95.6%),cow(Bos taurus-bosTau3) (91.8%),domesticated dog (Canis lupus familiaris-canFam2)(89.3%),rat (Rattus norvegicus-rn4) (82.4%),mouse (Mus musculus-mm9) (84.3%) and opossum (Monodelphis domestica-monDom5) (78.0%).But the region was not conserved in chicken (Gallus gallus),frog (Xenopus laevis),zebrafish (Danio rerio),fugu pufferfish (Takifugu rubripes) and spotted green pufferfish (Tetraodon nigrovoridis).An overview of the mammalian conservation of the in silico-predicted promoter ofTNFis shown in Figure 5.
Prediction of TFBSs:In the present study,we used five software programs to predict TFBSs.To ensure accurate analysis,only factors predicted by three out of the five programs or predicted by two programs but verified in the literature were selected.The five prediction programs reported multiple putative TFBSs within the (-146 to+10) region,as illustrated in Table 5.Moreover,screening of this region using the NCBI SNP databases showed four SNPs,as shown in Table 5.The ECR Browser and NCBI Basic Local Alignment Search Tool nucleotide revealed the conservation of these SNPs in chimpanzees,rhesus monkey,cow and dog.Multiple-sequence local alignment and visualization presented multiple TFBSs to be located at rs765073823 and rs537401710.The overview from the multiple-sequence local alignment and visualization software for conserved TFBSs predicted to be conserved (100%) between humans,chimpanzees,rhesus monkeys,cows,and domesticated dogs is summarized in Table 6.
Prediction of CEs:In the present study,CEs were predicted using MatrixCatch,which can find experimentally known regulatory elements (both single sites and pairs) and novel regulatory elements by computational comparison to the known ones in a library of CE models[51].An overview of predicted CEs by MatrixCatch is presented in Table 7.
Allele and genotype frequencies of TNF-A-1030 and susceptibility to H.pylori infection
Sixty-one participants from each group (H.pylori-infected and uninfected) were successfully genotyped forTNF-A-1030 polymorphism;a representative gel is shownin Figure 6.Allele and genotype frequencies ofTNF-A-1030 polymorphism in patients infected withH.pyloriand uninfected controls are presented in Table 8.Although the TNF-A-1030-C allele was more prevalent in patients infected withH.pyloricompared to uninfected controls,no significant association was found between the allele frequencies ofTNF-A-1030 andH.pyloriinfection (27.87%vs19.67%,OR=0.6339,95%CI:0.3490-1.151,P=0.176).However,a significant association was detected between T/C genotype andH.pyloriinfection (39.34%vs19.67%,OR=2.69,95%CI:1.17-6.17,P=0.020) (Tables 8 and 9).In contrast,no significant association was observed between allele and genotype frequencies ofTNF-A-1030 and gastric cancer (P=0.8116 andP=0.3900,respectively) (Table 10).The allelic and genotype distributions at -1030 ofTNF-Awere in Hardy-Weinberg equilibrium,with non-significantχ2values(for uninfected subjectsP=0.1520;and subjects with benign disordersP=0.2089).
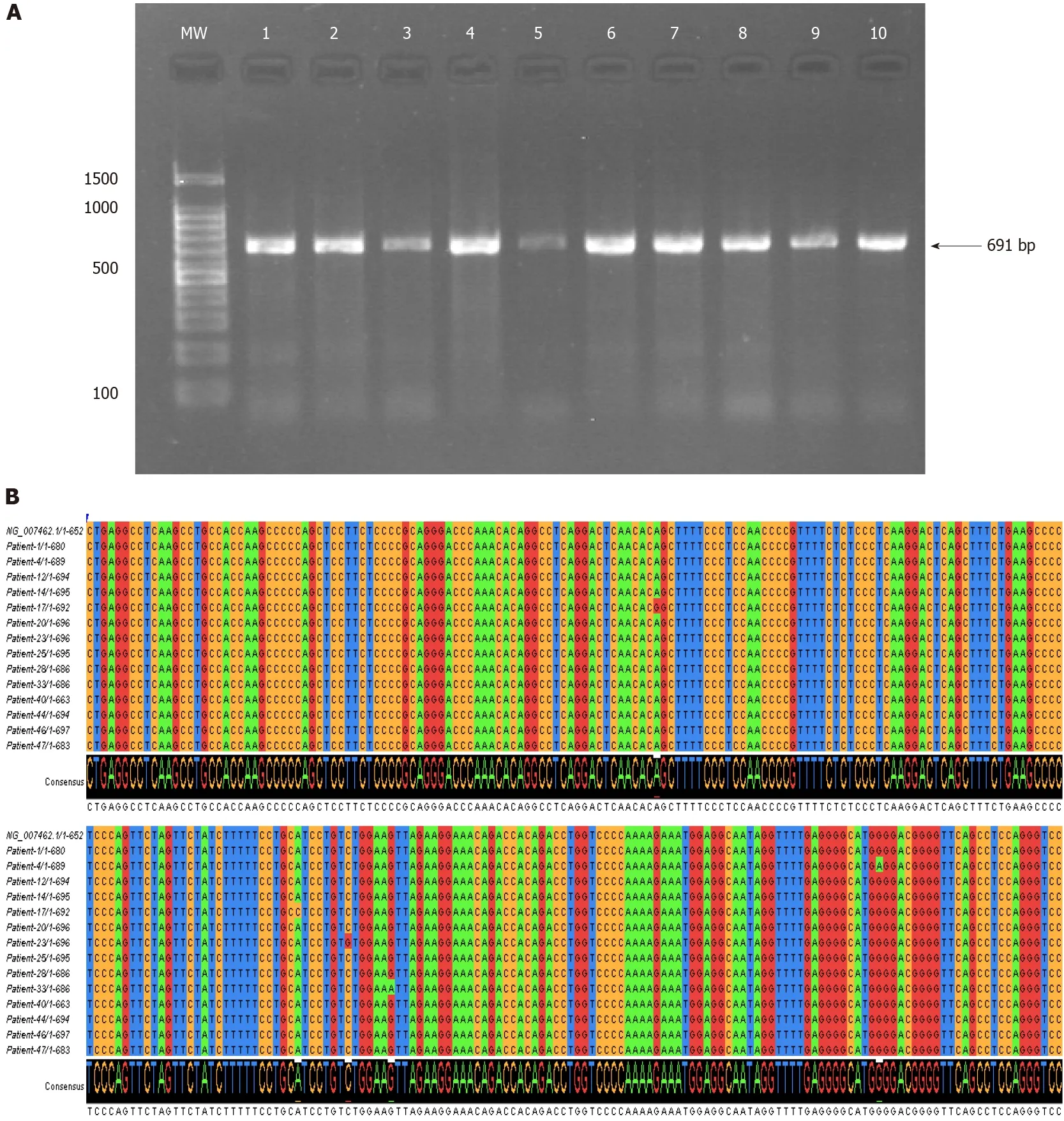
Figure 2 Nucleotide variations of the tumor necrosis factor-alpha 5’-region (-584 to +107) in the Sudanese Helicobacter pylori-infected patients.A:PCR amplification results of the tumor necrosis factor-alpha 5’-region (-584 to +107) examined on 1% agarose gel electrophoresis;B:Multiple sequence alignment of the nucleotide sequences of the tumor necrosis factor-alpha 5’-region performed in Clustal W and visualized in Jalview[55].The levels of identity are visible as histograms at the bottom.

Table 3 Nucleotide variations in the 5’-regulatory (-584 to +107) region of tumor necrosis alpha in Sudanese Helicobacter pylori-infected patients

Table 4 Overview of the in silico predicted tumor necrosis factor-alpha promoter regions for the respective prediction programs.The most predicted region is indicated in bold

Table 5 Summary of the in silico predicted transcription factor binding sites for tumor necrosis factor-alpha (-146 to +10) region

Table 6 Overview of conserved transcription factor binding sites predicted by multiple-sequence local alignment and visualization
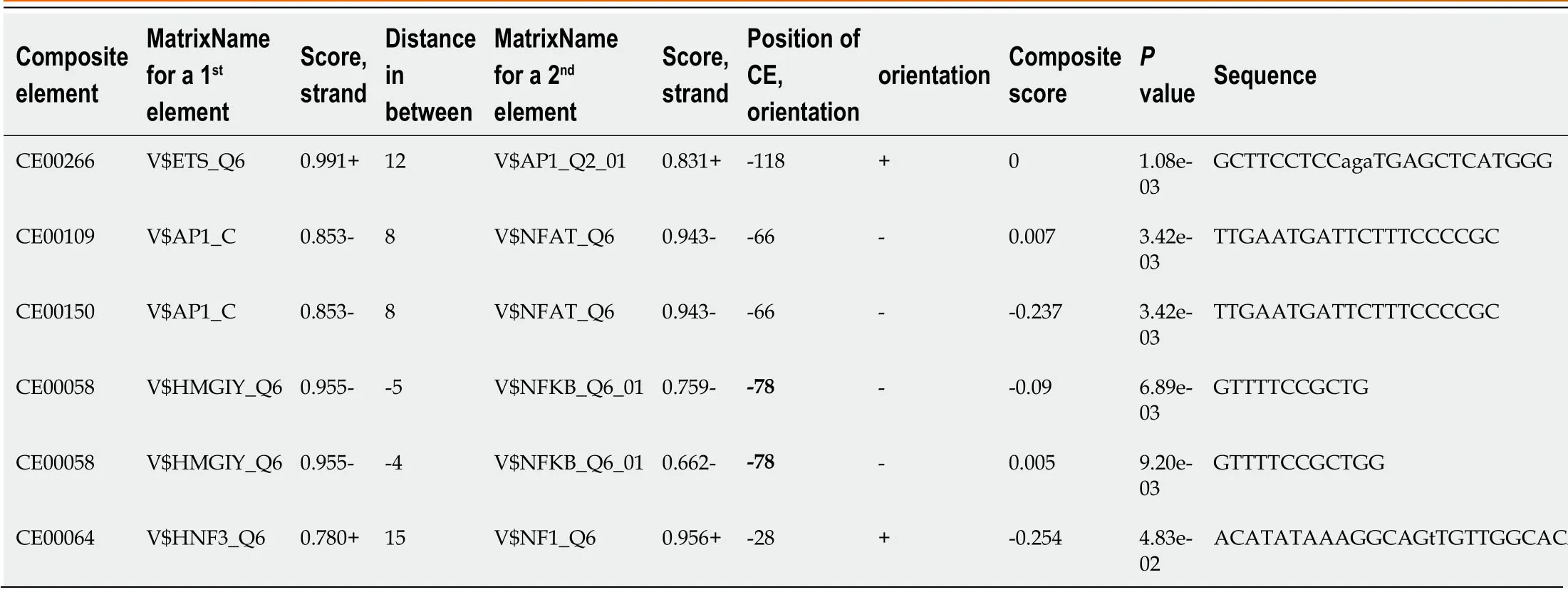
Table 7 Summary of the in silico predicted composite regulatory elements for the -146 to +10 region of tumor necrosis factor-alpha
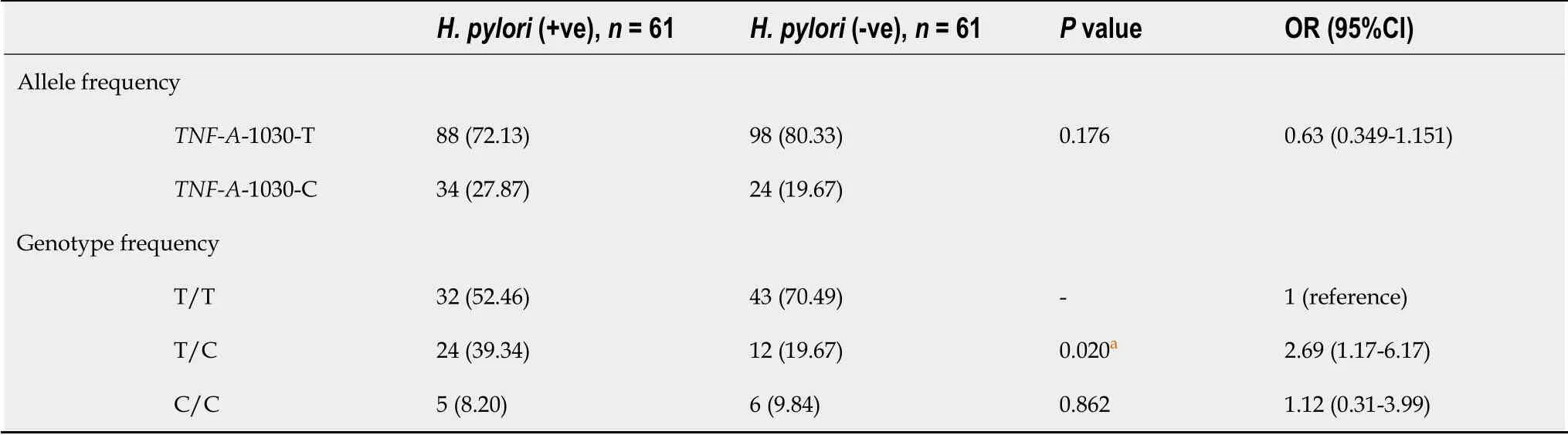
Table 8 Allele and genotype frequencies of tumor necrosis factor-alpha-1030 polymorphism among Helicobacter pylori-infected and uninfected subjects and their contributions to Helicobacter pylori infection,n (%)
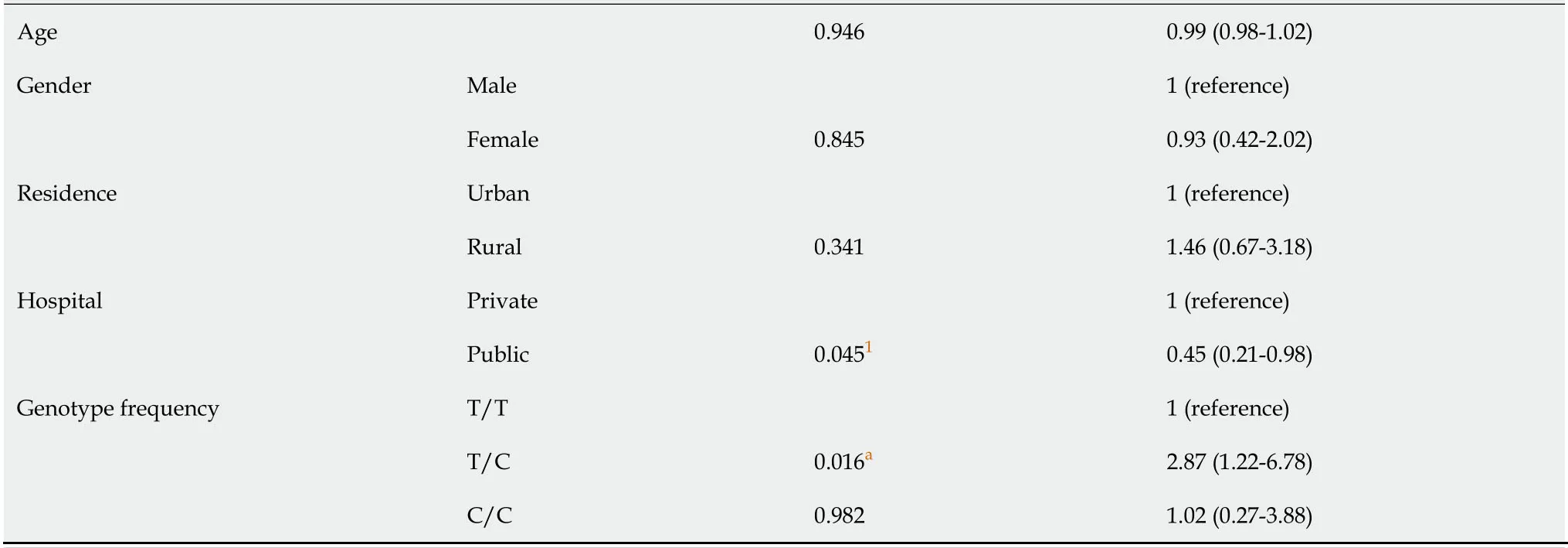
Table 9 Multivariate analysis of genotype frequencies of tumor necrosis factor-alpha-1030 polymorphism and sociodemographic characteristics in Helicobacter pylori infection

Table 10 Tumor necrosis factor-alpha-1030 polymorphism and gastric cancer association risk in gastric cancerous patients and subjects with benign gastric disorders,n (%)
DlSCUSSlON
Many SNPs have been observed in the vicinity of theTNFlocus,particularly in the distalTNFpromoter.Some of these have been implicated in the disease susceptibility,resistance or severity by modifying the transcriptional regulation ofTNF[13,56-58].In this study,we functionally analyzed SNPs in theTNF-A5’-region (-584 to +107) of the Sudanese patients infected withH.pyloriand had divergent clinical outcomes that ranged from simple asymptomatic gastritis to more serious conditions,i.e.peptic ulcer disease and gastric cancer.The exploratory sequencing ofTNF-A5’-region (-584 to+107) of the Sudanese patients infected withH.pylorirevealed six previously reportedSNPs located at -567,-375,-307,-237,-76 and +70 and one novel SNP at +27 relative to theTNF-Atranscription start site.A C to G homozygous transversion at -567 nt was found in all our studied populations.At the same time,the novel A to T heterozygous transversion at +27 nt was observed in five patients who had normal upper gastroendoscopy or gastritis.This SNP is located in the 5’ untranslated region.Therefore,it could affect the post-transcriptional regulatory pathways that control mRNA localization,stability and translation efficiency[59].Further studies are encouraged to investigate this SNP in terms of the frequency of the minor allele (T) in the Sudanese population and its functional significance using computational and experimentalapproaches.
Intriguingly,the heterozygous SNP T>A at -76 was located at the in silico-predicted promoter region.However,in this study,the in silico analysis predicted promoter region was located from -146 to +10 nt relative to the TNF-α mRNA cap site using different algorithms[26].This finding is in agreement with previous studies conducted on 411TNFpromoters from individuals of distinct ethnic backgrounds that concluded that the humanTNFpromoter sequences up to 200 bp relative to the transcription start site involved in enhanceosome formation and the regulation of the gene are completely conserved in humans[21,26,60,61].Also,the ENCODE data showed a high level of the hallmarks of cis-regulatory regions at the in silico predicted promoter region (-146_+10),which include nuclease hypersensitivity and histone modifications[40-42].
Functional elements in the mammalian gene promoters can be detected by high conservation between species distantly related on the evolutionary scale[62,63].In this study,the computational comparative analysis revealed mammalian conservation for the -146 to +10 region in chimpanzee (Pan troglodytes-pan-Tro2) (99.9%),rhesus monkey (Macaca mulatta-rheMac2) (93.7%),cow (Bos taurus-bosTau3) (78.2%),domesticated dog (Canis lupus familiaris-canFam2) (77.8%),rat (Rattus norvegicus-rn4)(76.3%),mouse (Mus musculus-mm9) (78.7%) and opossum (Monodelphis domestica-monDom5) (72.2%).In an earlier study conducted by Leunget al[64],the region from positions -131 to -63 and -53 and -45 relative to the start site ofTNFgene transcription showed high sequence conservation among primates,with complete conservationwithin phylogenetic footprints across seven primate species,including Old World monkeys and New World monkeys[64].
However,theTNF-Agene is located within the major histocompatibility complex region of chromosome 6,the highly polymorphic region of the human genome,which includes the human leukocyte antigen (HLA) genes[65].In fact,all of the common ten detected SNPs (-1030,-862,-856,-574,-375,-307,-243,-237,and +70 nt relative to the start site of transcription) fall outside of the identifiedTNFprimate phylogenetic footprints[66-70].This feature is consistent with the accrual of mutations under neutral evolution[64,72].Furthermore,several of these SNPs (-1030,-862,-856,-307,-243,-237)have been shown to be in non-random association with extended HLA haplotypes[21,26,71-73],which are HLA regions that exhibit fixity of the genomic interval between HLA-B and HLA-DR,that includesTNF-Aand complement (complotype) regions[21,74,75].
Intriguingly,we detected one mutation (T>A,-76) located at the identifiedTNFprimate phylogenetic footprints[64] in a patient with reinfection withH.pylorimultiple times.The presence of this SNP in the conserved region that is involved in regulating theTNFgene could affect the expression and create a condition of hypoacidity that favors the survival and colonization ofH.pylori[76,77].The -76 SNP was first discovered with an overall minor allele frequency of 0.031 in the Indian population while studying polymorphisms ofTNF-enhancer and the gene for FcγRIIa correlated with the severity ofFalciparum malariain the ethnically diverse Indian population[78].It is present at the low-affinity transcription factor Ets-1 binding site in macrophages and the high affinity NFATp binding site in T and B cells[79].Also,it is located at an in silico-predicted C/EBP-beta (also known as NF-IL6) binding site (in this study,see Table 5),which was experimentally proven in myelomonocytic cells[80-82].
Sequence analysis of theTNF-A5’-region (-584 to +107) of the Sudanese patients revealed relative variability ofTNF-ASNPs (Figure 2 and Table 3).This finding is in agreement with a study conducted by Baenaet al[21] that found that the highest relative diversity ofTNF-ASNPs was revealed among the three African studied populations (Malawian,Southern Nigerian,African-American and African-Caribbean).Also,it is consistent with the“out of Africa”hypothesis based upon human mitochondrial DNA analyses[83].Despite this high level of variation,here in this study,the sequences involved in enhanceosome formation and gene regulation are highly conserved[64].Remarkably,we detected only a single SNP (-76) in this region.By contrast,the novel +27 variant and otherTNF-ASNPs described in this study occur within or immediately adjacent to promoter regions with high diversity in the primate lineage.Unexpectedly,the -243 variant,the firstTNF-ASNP linked to an Africanderived extended HLA haplotype[70],was not detected in our studied population.Other previously identified SNPs in this region (-584 to +107),which include -567,375,-307,-237,-76 and +70 nt relative to the start site of transcription,were observed[66-68,84].

Figure 3 MethPrimer software prediction of no CpG islands in the in silico predicted promoter region.

Figure 4 Overview from the ECR Browser shows mammalian conservation of the tumor necrosis factor 5’-upstream region compared to the human sequence in the region (-584 to +107) (hg19 chr6:31542751-31543444).Blue boxes represent the first tumor necrosis factor exon,while yellow indicates the tumor necrosis factor 5’ untranslated region.Intragenic positions are highlighted in red or in green when corresponding to transposable elements and simple repeats.UTR:Untranslated region.

Figure 5 Mammalian conservation of (AT;single nucleotide polymorphism databases dbSNP:rs41297589) at position -76 and the novel mutation (A>T at +27) in the 5’ untranslated region among various species.The nucleotides are enumerated at each line on the right side,and the in silico predicted TATA-,C/EBP-beta and transcription start site have marked inboxes.The chromatogram results of the polymorphisms are visualized using Finch TV software.TSS:Transcription start site.

Figure 6 Molecular detection of the tumor necrosis factor-alpha-1030 C/T polymorphism.A:PCR with confronting two-pair primer products analyzed on a 1% agarose gel stained with ethidium bromide.Three genotypes can be seen.Lanes 2,3 and 5 showed 444 bp,316 bp and 174 bp,indicating a heterozygous genotype.Lanes 4 and 6 showed 444 bp and 316 bp,which indicated a homozygous T genotype.Lane 1 showed 444 bp and 174 bp,which indicated a homozygous C genotype;B:Mammalian conservation of (TC;dbSNP:rs1799964) position -1030 among different species.MW:Molecular weight marker;C-ve:Negative control.
The -1030 (T/C;rs1799964) SNP is frequently observed in African populations (17%-25% Malawian,7% Nigerian,29% African-American/Caribbean and 14% Gambia)[21,22].In this study,the -1030 SNP was detected in 47 Sudanese (38% of our studied population).Many studies have been published that examine the association between the -1030 SNP and multiple diseases[60,85,86] and diseases related toH.pylorisuch as gastroesophageal reflux disease,gastritis,peptic ulceration and gastric cancer[87-90].In this study,we found a lack of association between -1030T and susceptibility toH.pyloriand gastric cancer in the studied population (P=0.1756 andP=0.8116,respectively).This finding is in agreement with previous studies[87,88,91].In contrast to a study conducted in Japan that observed a significant association between -1030T and gastric cancer[91].This variation could be attributed mainly to differences in genetic backgrounds of the studied population,the method of genotyping and sample size[76,92].
The study’s limitations include the relatively small number of the enrolled subjects and depending on the in silico tools for studying the influence of the detected promoter variants on theTNF-Agene expression andH.pyloriinfection (susceptibility and progression).Further large cohort studies are needed to validate the study findings.However,the computational analysis provides a basis for identifying promoter regions,recognizing regulatory motifs and understanding gene expression patterns[25].
According to the literature,a considerable number of polymorphisms have been discovered in theTNF-Apromoter.However,a crucial question that remains to be answered is whether these polymorphisms have any functional effect that may significantly impact disease incidence or severity.Identifying which of these many variants are functional is of great significance for discovering new preventive,diagnostic and therapeutic strategies against the incidence and/or progression of multifactorial diseases such asH.pyloriinfection.This study detected seven SNPs in theTNF-A5’-region;only one of them (T>A,-76) was located at the in silico-predicted promoter region (-146 to +10).This SNP could lead to the modification of the transcriptional regulation ofTNF-AinH.pyloriinfection.However,this conclusion cannot substitute for the experimental proofs (in vitroorin vivo),but it can provide a direction or insight for such experiments to validate the in silico predictions.
CONCLUSlON
In the SudaneseH.pylori-infected patients,seven SNPs were observed in theTNF-A5’-region;only one of them (T>A,-76) was located at the in silico-predicted promoter region (-146 to +10).In addition,it was predicted to alter TFBSs and CEs.Furthermore,a novel mutation (A>T,+27) was detected in the 5’ untranslated region,and it could affect the post-transcriptional regulatory pathways.In addition,computational analysis was a valuable method for understanding gene expression patterns and providing guidance for furtherin vitroandin vivoexperimental validation.
ARTlCLE HlGHLlGHTS
Research background
Helicobacter pylori (H.pylori) infection represents a major public health challenge in Sudan.However,functional polymorphisms within the tumor necrosis factor-alpha (TNF-A) promoter are associated with the incidence and progression of H.pylori infection by increasing TNF-a.production.
Research motivation
TNF lies within the major histocompatibility complex region of chromosome 6,which is a highly polymorphic region.Therefore,many polymorphisms have been detected in the TNF-A promoter and studying which variants could affect TNF-A gene expression is relevant.However,the influence of these single nucleotide polymorphisms (SNPs) on TNF-a production is not fully known and still a contradictory topic of debate due to the ethnic differences between populations.Furthermore,to our knowledge,there are no previous studies in Sudan that have addressed the association between TNF-A and H.pylori infection or H.pyloriassociated diseases.
Research objectives
To functionally characterize the genetic variations in the TNF-A 5'-region (-584 to+107) of Sudanese patients infected with H pylori and predict if these SNPs could alter the regulatory motifs using bioinformatics analyses.Also,to investigate the mammalian conservation of these SNPs using comparative profiling analysis in 11 species.
Research methods
An observational study was conducted in the major hospitals in Khartoum state.Genomic DNA was extracted from 122 gastric biopsies of patients who had been referred for endoscopy.Genotyping of the TNF-A-1030 polymorphism was performed using PCR with confronting two-pair primer to investigate its association with H.pylori infection in the Sudanese population.Sanger sequencing was applied to detect SNPs in the 5'-region (-584 to +107) of TNF-A in H.pylori-infected patients;in silico tools were used to predict whether these mutations would alter transcription factor binding sites or composite regulatory elements in this region.In addition,the ECR browser and multiple-sequence local alignment and visualization search engine were used to study the conservation of the detected SNPs among 11 mammalian species.
Research results
A total of seven SNPs were observed in theTNF-A5’-region of Sudanese patients infected withH.pylori.Among them,the SNP (T>A,-76) was located at the in silicopredicted promoter region (-146 to +10),and it was predicted to alter transcription factor binding sites and composite regulatory elements,while the novel mutation(A>T,+27) was detected in the 5’ untranslated region.It could affect the posttranscriptional regulatory pathways.Mammalian conservation was detected for the (-146 to +10) region in chimpanzee (99.4%),rhesus monkey (95.6%),cow (91.8%),domesticated dog (89.3%),mouse (84.3%),rat (82.4%) and opossum (78.0%).Furthermore,genotyping ofTNF-A-1030 revealed a lack of significant association between -1030T and susceptibility toH.pyloriand gastric cancer in the studied population (P=0.1756 andP=0.8116,respectively).
Research conclusions
Despite the high level of genetic variation in theTNF-A5’-region (-584 to +107) of the Sudanese patients,the sequences involved in enhanceosome formation and gene regulation are highly conserved.Remarkably,only a single SNP (-76) was detected in this region.In addition,computational analysis was a valuable method for studying gene expression patterns and insights for furtherin vitroandin vivoexperimental proofs.
Research perspectives
Further large cohort studies are needed to assess the association between (T>A,-76)mutation andH.pyloriinfection (susceptibility and progression).Also,further studies are encouraged to investigate the novel mutation (A>T,+27) in terms of the frequency of the minor allele (T) in the Sudanese population and its functional significance using computational and experimental approaches.Identifying which of these detected variants are functional is of great relevance for discovering new preventive,diagnostic and therapeutic strategies against the incidence and/or progression of multifactorial diseases such asH.pyloriinfection.
ACKNOWLEDGEMENTS
We gratefully acknowledge the patients and the staff of the endoscopic unit in Soba teaching hospital,Ibin Sina specialized hospital,Police hospital,Modern Medical Centre,and Al Faisal Specialized Hospital.Also,we would like to show our gratitude to the Department of Molecular Research Laboratory at the Faculty of Medical Laboratory Sciences,the University of Khartoum,for their cooperation.
 World Journal of Gastroenterology2022年2期
World Journal of Gastroenterology2022年2期
- World Journal of Gastroenterology的其它文章
- Nanotheranostics:A powerful next-generation solution to tackle hepatocellular carcinoma
- Multiple subcellular localizations and functions of protein kinase Cδ in liver cancer
- Prospective Study Outreach onsite treatment with a simplified pangenotypic directacting anti-viral regimen for hepatitis C virus microelimination in a prison
- Therapeutic endoscopy for the treatment of post-bariatric surgery complications
- Update on the applications and limitations of alpha-fetoprotein for hepatocellular carcinoma
- Case Control Study Obesity is associated with decreased risk of microscopic colitis in women