
临床研究样本量的估计方法和常见错误
2022-02-14 02:26潘岳松金奥铭王梦星
中国卒中杂志 2022年1期
潘岳松,金奥铭,王梦星
近年来,随着我国对临床研究的重视,国内脑血管病领域的相关研究迅速发展。样本量估计是临床研究设计中的核心问题之一,随着临床研究的发展,也越来越受到关注和重视。最近的一项统计显示,N Engl J Med发表的文章中,使用样本量计算/统计功效分析的在1978-1979年只有39%,而在2015年增长到了62%,是目前该杂志发表文章中使用频率最高的统计学方法[1]。样本量的正确估算直接关系到临床研究的成败,也一直是临床研究者较为关注且难以把握的关键点之一。如果样本量太小,研究结果的可重复性及代表性就欠佳,可能得出假阴性或假阳性的结论;如果样本量过大,研究所需的经费和资源就越多,项目执行的难度也越大,而且试验过程中对研究对象可能的潜在伤害越大,还存在伦理问题。那么,临床研究设计时如何确定合适的样本量呢?本文对决定样本量估计的要素、常用的脑血管病临床研究样本量计算方法、样本量估计中常见的问题等方面进行阐述,以促进脑血管病领域的研究者更好地掌握临床研究样本量估算的方法。
1 决定样本量估计的要素
1.1 研究目的、研究设计和主要观察指标的资料类型 临床研究的样本量估计首先需考虑研究目的和研究设计类型,根据不同的研究类型来选择对应的估计方法[2]。首先应明确研究的目的是分析疾病发病或预后的危险因素,是验证某项干预措施的有效性和安全性,还是评估某项新技术诊断疾病的准确性。研究目的不同,对应的样本量估计的考虑因素也不同。临床研究设计类型是属于病例对照研究,还是队列研究、随机对照试验?随机对照试验是优效性设计,还是非劣效性设计?暴露因素/干预措施分组是两组还是多组?是否匹配设计?不同的临床研究设计类型对应的样本量估计需考虑的因素和计算公式各不相同。在估计样本量之前,首先要明确和充分理解研究的目的和研究设计的类型。
样本量估计需考虑的另一个重要因素是主要观察指标的资料类型。主要观察指标的资料类型一般可以分为定性指标和定量指标两种,对应的样本量估计方法各不相同。脑血管病临床研究中常用于评估二级预防效果的结局指标如卒中复发、联合血管事件发生、缺血性卒中复发等;常用于溶栓、机械取栓、神经保护等临床试验的结局指标如功能预后良好(mRS 0~1分或mRS 0~2分);Ⅱ期临床试验常用的神经功能恶化/好转等指标均为二分类定性指标。对主要观察指标为二分类定性指标的临床研究,样本量估计主要采用率的比较计算公式,用到的主要参数为率。常用于卒中后认知功能障碍、脑小血管病预后研究的认知功能评分等指标为定量指标,对主要观察指标是定量指标的临床研究,样本量估计主要采用均数比较的计算公式,用到的主要参数为观察指标的均数和标准差。
1.2 效应值 暴露组或干预组间的主要观察指标的预估效应值大小是直接决定临床研究所需样本量最主要的因素之一。对于两组比较,以Δ表示两组总体参数(均数或率)的差值或比值。常用的效应值包括组间MD、RD、RR、HR、OR等。一般来说,两组的预估效应值越大,如两组MD或RD越大,RR、HR或OR越偏离1,所需的样本量越小。当临床研究是优效性试验或非劣效性试验设计时,还需结合比较优效性界值或非劣效性界值来确定样本量。
1.3 变异度 一般用方差或标准差反映组间观察指标的总变异程度。两组定量指标(均数)的比较,其方差可通过两组样本方差估计。两组定性指标(率)的比较,其方差可通过两组样本率估计。一般情况下,变异度,即方差越大,所需样本量越大。事件率越接近0.5,所需样本量越小。
1.4 检验水准 检验水准α,即Ⅰ型错误的概率,是指错误地拒绝了实际成立的原假设H0,错误地判定为有差异的概率大小。α越小,所需样本量越大。α的取值常为双侧0.05或0.1,优效或非劣效试验设计中常取值为单侧0.025。
当需要多重检验时,如设置了多个主要疗效指标、拟进行多组间两两比较或在试验过程中设计了期中分析,需进行多次比较分析的情况下,则会使Ⅰ型错误增加,需对α进行校正。在这一过程中要进行多次重复显著性检验,每进行一次检验都将增加Ⅰ型错误的概率,从而使总的显著性水平α上升。如,以检验水准α=0.05,重复进行10次检验为例,发生Ⅰ型错误的总概率将上升到0.19。常用的调整检验水准α的方法,如Bonferroni法,调整后的α’=α/k(k为统计检验的次数),如总共需进行3次检验,则α’=0.05/3,为0.0167。成组序贯研究设计包含了期中分析,为了使总体显著性水平维持常数α,必须调整每一次分析的显著性水平,常用的调整α水平的方法包括Pocock法、O’Brien-Fleming法和Peto法等[3]。
1.5 把握度 把握度,即检验效能,是指所研究对象总体间确有差异时,按检验水准α能够发现此差异的概率。把握度=1-β,其中β为Ⅱ型错误的概率,因此指定了β水平也就等于指定了把握度水平。把握度越大,所需样本量越大,通常将其定为0.80或0.90。一般建议临床试验把握度定为0.90。
1.6 其他因素 除了上述主要因素外,其他因素如两组例数的分配比例、优效性与非劣效性界值、不应答或失访率等均可影响样本量的估计[4]。一般而言,两组比较时取相等的样本含量,此时总的样本含量最少,且可达到最高的统计效能,因此经常使用的是各组等样本含量设计。但是,由于某些实际原因,有时可能需要进行各组不等样本含量设计,在进行样本量估计时也应予以考虑。通过样本量估算公式计算得到样本量后,一般要考虑不应答或失访的影响,增加相应的样本量,以确保实际收集的有效病例数能足够达到统计要求。如考虑失访的影响,可将最后的样本量定为N’=N/(1-失访率)。
2 样本量估计的思路
不同的临床研究设计样本量估计方法也不同,样本量估计的思路非常重要(表1)。与临床研究样本量估计相关的最核心的参数是“两组主要观察指标的预估值”。在根据样本量估计公式计算之前,需要先明确3个问题:①研究的目的是什么?采用什么样的设计?②研究的主要观察指标是什么?③主要观察指标有预估值吗?其中,研究目的、研究设计和主要结局指标在研究方案设计时应该就已经确定了。而主要观察指标的预估值,则可通过预试验或总结前期数据、查阅文献及结合专家意见,由临床专家和统计学专家联合确定。确定了上述3个问题的答案,就确定了研究样本量估计时需要用到的参数及参数的大小,之后将数据代入样本量计算公式,采用软件计算即可。常用的PASS、SAS、Stata等统计软件以及一些公众网站和微信小程序都可以方便地实现样本量的计算[5]。

表1 样本量估计的思路
3 常见的临床研究设计样本量估计方法
3.1 临床登记队列样本量估计 在脑血管病临床研究中,最常见的研究设计类型是临床专病队列,用于探讨基线预后影响因素与患者预后的关系。根据危险因素的有无或高低可分为两组,进而比较两组卒中复发率的差异或检验暴露因素与患者预后的关系。两样本率比较的样本量估计可采用公式①进行计算。其中p1与p2分别代表两组的率,p=(p1+p2)/2。有时实际得到的是对照组的率p1和效应值RR或OR值,可通过公式p2=RR×p1或公式p2=(OR×p1)/[1+p1×(OR-1)]进行转化。Z1-α/2和Z1-β分别为α和1-β对应的标准正态分布临界值。实际工作中,可进一步考虑两组不等比例、混杂因素的影响等对样本量做进一步调整,扩大样本量。

示例:某研究者拟进行一项临床登记研究探讨基线糖尿病状态与卒中患者1年卒中复发的关系。前期资料显示糖尿病的RR值为1.3,无糖尿病人群的一年卒中复发率为8%,取α=双侧0.05,β=0.20,考虑失访率为10%,计算得所需样本量为2529例/组。
3.2 优效性临床试验两样本率比较 优效性临床试验的目的是评价试验干预措施是否优于对照措施(阳性或安慰剂对照)。如双联抗血小板治疗对比单联抗血小板治疗的试验、机械取栓对比传统治疗的试验均多采用这种研究设计[6-7]。两样本率比较优效性临床试验的样本量估计可采用公式②进行计算。其中p1与p2分别代表两组率,p=(p1+p2)/2,Δ为优效性界值。Δ一般取具有临床意义的最小值,由临床专家与统计学专家协商确定。当Δ=0时,优效性检验相当于两样本率比较的样本量计算方法。
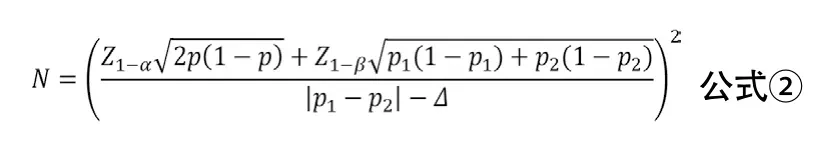
示例:某试验拟比较阿司匹林联合氯吡格雷双联抗血小板治疗与单用阿斯匹林治疗对预防卒中复发的疗效,根据前期的文献报告,预设阿司匹林联合氯吡格雷双联抗血小板治疗组3个月卒中复发率为8%,单用阿斯匹林治疗组3个月卒中复发率为10%,优效性界值Δ=0,α=0.025,β=0.10,考虑失访率为5%,计算得所需样本量为4528例/组。
3.3 非劣效性临床试验两样本率比较 非劣效性临床试验的目的是评价试验干预措施在临床意义上不差于(非劣于)对照措施(通常为已上市的有效药物或标准治疗方案),目的是探索新的治疗选择。这种情况常用于原有上市药物的疗效较好,试验药的疗效超过标准治疗措施的可能性较小,预计研究药物的疗效与原有上市药物相当,但研究药物可能具有其他特点,在其他方面可能有优势,如更好的安全性、使用更方便或依从性更好等。随着药物研发的深入,近年来采用非劣效性设计的试验越来越普遍。脑血管病临床研究中如比较低剂量对比标准剂量阿替普酶溶栓、直接取栓对比桥接取栓、取栓前替萘普酶对比阿替普酶溶栓等试验多采用这种设计[8-10]。
两样本率比较的非劣效性临床试验的样本量估计可采用公式③进行计算。其中p1与p2分别代表两组率,p=(p1+p2)/2,Δ为非劣效性界值。非劣效性临床试验通常设两组的率相等。Δ一般由临床专家与统计学专家根据既往研究证据结合临床意义共同确定,并最终由临床专家确认。统计上可采用两步法估算,先估计出阳性对照药物相对于安慰剂为对照的绝对疗效M1,一般取小于阳性对照与安慰剂效应之差的95%CI下限(高优指标)[11]。临床可接受的非劣效性界值M2一般通过M2=f×M1计算确定,建议非劣效设计中取f=0.5。在没有历史数据可依据时,Δ的确定也可根据目标值法取值为阳性对照药物疗效的10%~15%。

示例:某试验采用非劣效性试验设计,拟检验直接取栓治疗患者3个月预后良好的比例不劣于桥接取栓治疗的患者。根据前期数据和文献报告,预设两组患者3个月预后良好率为60%,非劣效性界值Δ=5%,α=0.025,β=0.10,考虑失访率为5%,计算得所需样本量为2124例/组。
以上为脑血管病临床研究中常见的样本量估计方法,特殊类型研究,如整群随机对照临床试验、适应性设计临床试验、单组目标值试验、诊断性试验等的样本量计算,以及两样本均数比较的样本量估计方法,可参阅相关文献,因为篇幅的限制,本文不作详述。
4 样本量估计的常见错误
4.1 没经过计算直接确定样本量 部分研究者在撰写临床研究方案时,未经过计算就直接确定样本量(如100例或200例)。这种确定样本量的做法可能导致样本量不足,达不到统计学检验的要求,得不到预期的研究结果。不过,对于因实际情况限制无法入组过多的研究,或研究本身的目的是为了进行预试验探索方案的可行性、初步探索干预措施的疗效和安全性,可不按照样本量估计的例数入组,但应对预试验的研究目的给予明确说明。
4.2 样本量估计方法与研究设计和主要观察指标不对应 在样本量估计过程中,最常见的错误之一是样本量估计方法与研究设计和主要观察指标不对应。如:研究的目的是采用队列研究设计验证基线时某指标升高与卒中预后的关系,但样本量估计时却依据1年卒中复发率,采用率的横断面调查公式的方法进行样本量估计;或者,临床试验中设置的主要观察指标为3个月预后良好的比例,但因为缺少前期数据,主要观察指标无法预估,在样本量估计时采用了3个月卒中复发率作为计算的依据。样本量的估计方法应与研究目的、研究设计和主要观察指标相对应,否则无法得到预期的效果。
4.3 参数设置不合理 样本量估计的另一个常见错误是样本量估算的参数设置缺乏依据,或参数设置不合理、不符合临床实际情况。如:为节省样本量有意夸大事件率或预期的效应值,或设置的脱落率和失访率过高,设置的参数明显不符合临床的实际情况。上述情况都可能导致样本量估计不准确,从而使研究达不到预期的研究效度。
总之,在临床研究的过程中,样本量既不是越大越好,也不是越小越好。合理的样本量是临床研究科学设计的重要环节,与研究设计的其他环节密切相关,估计过程应充分理解和考虑研究目的、研究设计和主要观察指标的资料类型。样本量需要临床专家和统计学专家合作讨论确定,选择正确的计算方法和公式,合理设置参数,并进行科学的计算才能保证其准确无误。
猜你喜欢
心理学报(2022年10期)2022-10-12
中原商报·科教研究(2022年1期)2022-05-13
海外星云(2021年9期)2021-10-14
中国新闻周刊(2021年24期)2021-07-19
现代仪器与医疗(2021年1期)2021-06-09
大众健康(2020年7期)2020-08-25
爱你(2019年13期)2019-11-14
数学学习与研究(2019年10期)2019-07-02
新一代(2018年17期)2018-02-26
科技资讯(2016年27期)2017-03-01
