
硝苯地平缓释片在中国健康受试者体内的生物等效性研究*
2022-02-13 05:55余恒毅区淑蕴刘东梁标志高永坚任秀华
医药导报 2022年2期
余恒毅,区淑蕴,刘东,梁标志,高永坚,任秀华
(1.华中科技大学同济医学院附属同济医院药学部,武汉 430030;2.华中科技大学同济医学院附属同济医院Ⅰ期临床试验研究室,武汉 430030;3.国药集团广东环球制药有限公司,佛山 528305)
硝苯地平是具有代表性的钙拮抗剂药物,为治疗心绞痛和高血压的首选药物之一,临床上通常使用缓控释制剂[1]。前期研究以日本拜耳药品株式会社生产的Adalat®-L10为参比制剂(R),对国药集团广东环球制药有限公司已上市的硝苯地平缓释片(Ⅰ)T1进行质量一致性评价,体外释放度研究结果表明制剂T1与参比制剂R体外释放行为不一致,修改处方工艺后的制剂T2与参比制剂R体外释放曲线一致[2]。笔者在本文就该公司已上市的硝苯地平缓释片(Ⅰ)T1、修改处方工艺后的制剂T2与参比制剂R在中国健康受试者体内的药代动力学及生物等效性进行研究。
1 材料与对象
1.1材料
1.1.1试药 硝苯地平缓释片(Ⅰ)(受试制剂T1,国药集团广东环球制药有限公司,批号:140101,规格:每片10 mg);变更处方工艺后硝苯地平缓释片(Ⅰ)(受试制剂T2,国药集团广东环球制药有限公司,批号:20140201,规格:每片10 mg);硝苯地平缓释片(参比制剂R,商品名:Adalat®-L10,日本拜耳药品株式会社,批号:JPR4392,规格:每片10 mg);硝苯地平对照品(中国食品药品检定研究院,批号:100338-201404,含量:99.0%);硝苯地平-d6对照品(内标,加拿大Toronto Research Chemicals公司,批号:457002,含量:98%);HPLC级乙腈、甲醇购于美国TEDIA公司,水为实验室制备超纯水。
1.1.2仪器 LC-20AD型高效液相色谱仪(日本岛津)。API4000 Plus三重四级杆质谱仪(美国爱博才思)。MS105DU型电子分析天平(瑞士梅特勒-托利多)。Vortex-2 GENIE混合器(美国Scientific Industries)。Neofuge 13R型高速冷冻离心机(香港力康)。Scientific Heraeus Multifuge X1低温台式离心机(德国赛默飞世尔)。Elga Purelab Felx 3型超纯水机(英国ELGA);DW-86L626型立式超低温保存箱(青岛海尔)。
1.2受试者选择 本试验研究方案与知情同意书等研究文件获得华中科技大学同济医学院伦理委员会批准,所有受试者自愿签署知情同意书。
1.2.1入组标准 ①男性;②年龄18~40岁;③体质量≥50 kg,且体质量指数在19~24 kg·(m2)-1;④试验前2周内未服用其他任何药物。
1.2.2排除标准 ①经生命体征检查、体格检查、实验室检查结果异常且有临床意义者;②既往有严重疾病史者;③筛选前3个月内参加过其他药物临床试验者;④对研究药物及同类药物过敏者;⑤有药物滥用史,嗜烟、嗜酒者;⑥试验前1个月内献血者;⑦研究者认为不能入组的其他情况者。
1.3试验入组与完成情况 空腹试验共筛选104名男性受试者,30例合格入组。其中2例脱落(1例于第1周期给药前主动退出试验、1例于第2周期给药前主动退出试验),28例完成试验,年龄(23.86±2.79)岁,身高(172.14±6.07)cm,体质量(63.96±7.13)kg,体质量指数(19.30~23.77)kg·(m2)-1。
餐后试验共筛选124名男性受试者,30例合格入组。其中1例脱落(第2周期给药前主动退出试验),29例完成试验,年龄(24.07±2.98)岁,身高(172.83±6.87)cm,体质量(64.69±7.28)kg,体质量指数(19.05~23.99)kg·(m2)-1。
2 方法与结果
2.1给药方案与血样采集 本研究采用单中心、随机、开放、三周期、三序列、三交叉单次给药试验设计。空腹试验和餐后试验各入组30名健康男性受试者。30名受试者随机分成3组(R-T1-T2、T1-T2-R和T2-R-T1组),每组10人,每周期分别在空腹或餐后状态下服用硝苯地平缓释片10 mg,清洗期为7 d。
受试者于每周期给药前1 d入住I期病房,统一进食晚餐后禁食过夜(空腹至少10 h),服药前2 h内禁水。空腹试验受试者在空腹状态下口服试验药物10 mg,温水200 mL送服。餐后组受试者在服药前30 min内完成高脂餐进食[每份标准餐含能量3352~4190 J(1 cal=4.19 J),其中蛋白质约628.5J、碳水化合物1047.5 J、脂肪2095~2514 J],在餐后状态下口服试验药物10 mg,温水200 mL送服。在服药后2 h内受试者禁水,服药后避免剧烈运动。服药后4 h和10 h后进食标准中餐和晚餐,试验期间受试者禁止吸烟、饮用酒类和咖啡类饮料。
研究人员在受试者服药前1 h、服药后2,4 h测量其生命体征(包括体温、脉搏、呼吸、血压),并密切观察可能出现的低血压反应(例如头晕、头痛、脸色苍白、直立性眩晕、四肢冷、心悸等)及其他不良反应,在出组时进行实验室检查,心电图检查。
采血点设置为每周期的0 h(给药前1 h)及给药后0.5,1,1.5,2,2.5,3,4,5,6,9,12,24 h,研究护士于避光条件下采集受试者肘静脉血约4 mL,将血样置于含肝素钠抗凝剂的真空采血管中轻柔颠倒混匀,置于低温离心机中离心10 min(4 ℃、2260×g)。分离血浆于冻存管中,置于-80 ℃超低温冰箱贮存待测。
2.2测定方法与样品处理 采用液相色谱-串联质谱法(LC-MS/MS)法。因硝苯地平光不稳定[3],溶液配制与血样处理相关操作均在黄光灯下进行(波长570~580 nm)。
2.2.1色谱条件 色谱柱:Dikma Diamonsil C18(2.1 mm×150 mm,5 μm);流动相A:水;流动相B:乙腈,梯度洗脱程序: 0~0.5 min,20%B;0.51~1.9 min,20%→90%B;1.91~3.0 min,90%B;3.01~3.1 min,90%→20%B;3.11~5.0 min,20%B;流速:0.36 mL·min-1;柱温:40 ℃;自动进样器温度:4 ℃;进样量:10 μL。
2.2.2质谱条件 离子源为电喷雾离子化源(ESI),正离子模式检测,毛细管电压5.5 kV,源温度550 ℃;Curtain gas 25 L·min-1,Gas1 40 L·min-1,Gas2 40 L·min-1。扫描方式为多反应监测:硝苯地平检测离子对为m/z347.2→315.1(DP 55V,EP 10,CE 12 eV,CXP 15 V);硝苯地平-d6(氘代内标)检测离子对m/z353.2→257.2(DP 55V,EP 10,CE 26 eV,CXP 10 V)。
2.2.3血浆样品处理 于1.5 mL棕色EP管中加入血浆样品200 μL、内标工作液20 μL(硝苯地平-d6:1200 ng·mL-1)和乙腈600 μL,涡旋混合1 min后,于4 ℃、12 000×g条件下离心10 min,取上清液200 μL进样分析。
2.3方法学考察与评价
2.3.1专属性 取6例健康志愿者的空白血浆,每例受试者的空白血浆分别不加硝苯地平与内标、仅加定量下限(LLOQ)浓度的硝苯地平、仅加内标,按照“2.2.3”项进行操作后进样、分析。结果见图1。在硝苯地平及其内标的离子通道及保留时间窗中(均为3.78 min),血浆内源性物质的峰面积均小于硝苯地平LLOQ峰面积的20%,小于内标峰面积的5%,显示血浆内源性物质对样品测定无干扰,方法专属性好。
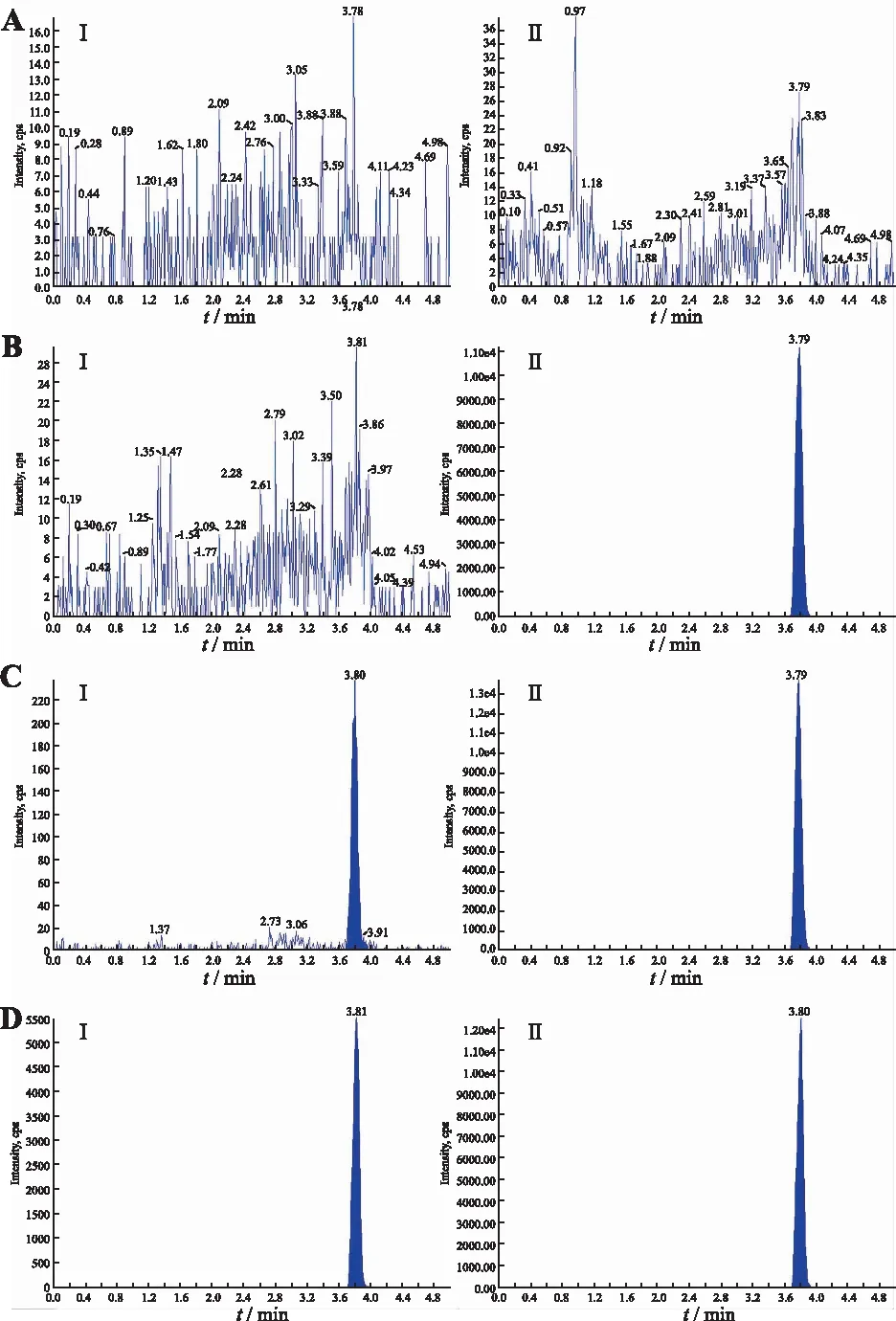
A.空白血浆样本(不加内标);B.空白血浆样本(加内标);C.定量下限(0.39 ng.mL-1)质控样本;D.健康受试者服药后样本;I为硝苯地平;II为硝苯地平-d6。图1 血浆中硝苯地平及硝苯地平-d6的典型色谱图A.blank plasma (without IS);B.blank plasma with nifedipine-d6 (IS);C.nifedipine at LLOQ (0.39 ng·mL-1) and IS;D.sample after administration of 10 mg nifedipine sustained-release tablet;I .nifedipine ; II nifedipine-d6.Fig.1 Typical multiple reaction monitoring chromatograms of nifedipine and nifedipine-d6
2.3.2标准曲线与定量下限 标准曲线血浆样品(0.39,1.56,3.90,7.80,39.00,78.00 ng·mL-1)按“2.2.3”项下处理后进样,以硝苯地平与内标峰面积之比为纵坐标(Y),以硝苯地平浓度为横坐标(X),采用加权(ω=1/X2)最小二乘法进行线性回归得回归方程为Y=0.039 4X-0.000 212(R2=0.997 3)。结果表明,硝苯地平在0.39~78.00 ng·mL-1范围内线性良好,定量下限为0.39 ng·mL-1。
2.3.3精密度及准确度 质控血浆样品(0.78,6.24,62.40 ng·mL-1)及定量下限血浆样品(0.39 ng·mL-1)按“2.2.3”项下处理后进样,于连续2 d内完成3次检测,每个浓度水平配制5个重复,使用随行标准曲线计算各质控样品及定量下限的浓度,计算批内、批间精密度及准确度,结果显示,定量下限及所有质控样品的批内及批间准确度均值在标示值的±15%范围内,批内及批间变异系数均在15%范围内[4]。
2.3.4提取回收率 质控血浆样品(0.78,6.24,62.40 ng·mL-1)按“2.2.3”项下处理后进样,每个浓度水平配制5个重复。配制含空白血浆样品提取液的化合物溶液使其浓度与质控样品进样浓度相同,比较两者峰面积比来评价化合物的提取回收率。结果显示,低、中、高3个质量浓度水平的提取回收率分别为(110.2±4.1)%、(91.9±3.0)%和(98.8±3.8)%,硝苯地平提取过程对真实结果影响较小。
2.3.5基质效应 分别配制同质控样品进样浓度相同的含空白血浆样品提取液的化合物溶液及同质控样品进样浓度相同的化合物溶液,进样测定后以两者经内标校正后的比值来比较硝苯地平的基质效应。结果显示,低、高浓度质控样品的校正后基质效应分别为99.9%和99.8%,变异系数在15%范围内,空白血浆中内源性物质不影响硝苯地平的测定。
2.3.6稳定性试验 取低、高质控血浆样品(0.78,62.40 ng·mL-1),每个浓度水平设5个重复,分别考察质控血浆室温放置24 h,-70 ℃条件冻融循环3次,及在-70 ℃条件下放置30,60 d的稳定性。质控血浆样品(0.78,62.40 ng·mL-1)按“2.2.3”项下处理后,置于仪器进样盘中24 h考察其稳定性。结果显示,每一浓度的均值与标示浓度的偏差在±15%范围内,表明硝苯地平血浆样品在室温、冷冻保存、反复冻融条件下稳定性良好。
2.4统计学方法 采用DAS 2.1软件,以非房室数学模型分析方法估算受试制剂和参比制剂的药动学参数,主要参数包括出峰时间(tmax)(实测值)、峰浓度(Cmax)(实测值)、血浆浓度-时间曲线下面积(AUC0-t)、AUC0-∞、和消除半衰期(t1/2)。
采用DAS 2.1软件进行生物等效性分析,对主要评价参数Cmax、AUC0-t、AUC0-∞进行对数转换后进行方差分析,然后用双单侧检验并计算90%置信区间(CI)。当受试制剂与参比制剂主要评价参数的几何均值比的90%CI在80%~125%等效区间内认为两制剂生物等效[5]。
2.5血药浓度-时间曲线 健康受试者空腹/餐后状态下单次口服硝苯地平缓释片受试制剂T1、受试制剂T2和参比制剂10 mg后,血浆中硝苯地平的平均浓度-时间曲线见图2所示。

图2 空腹(A)和餐后(B)状态下受试者单次口服硝苯地平缓释片受试制剂T1、T2与参比制剂R后平均血药浓度-时间曲线Fig.2 Mean plasma concentration-time curve of nifedipine after single oral test preparations T1,T2 and reference preparation R under fasting (A,n=28) or fed (B,n=29) state in healthy
2.6药动学参数 受试者空腹/餐后状态下单次口服硝苯地平缓释片受试制剂T1、受试制剂T2和参比制剂10 mg的主要药动学参数见表1。
表1 空腹和餐后状态下受试者单次口服硝苯地平缓释片受试制剂T1、T2与参比制剂R的主要药动学参数Tab.1 The main pharmacokinetic parameters of nifedipine in healthy volunteers after taking the tests T1,T2 and reference R preparations under fasting or fed state
2.7生物等效性评价 纳入生物等效性集的57例受试者空腹/餐后给予硝苯地平缓释片受试制剂T1、受试制剂T2和参比制剂10 mg的生物等效性评价结果见表2。如表2中所示, T1和参比制剂在空腹状态下的AUC0-t、Cmax生物不等效,在餐后状态下Cmax生物不等效。而改良后受试制剂T2和参比制剂在空腹及餐后状态下Cmax、AUC0-t、AUC0-∞的90%CI均在80%~125%内,符合生物等效性要求,判定为生物等效[5]。

表2 空腹和餐后状态下受试者单次口服硝苯地平缓释片受试制剂T1、T2与参比制剂R的生物等效性评价结果Tab.2 The parameter for the test preparations T1,T2 and reference preparation R of sustained-release tablets taken by healthy subjects under fasting and fed state
2.8安全性评价 整个试验期间未发生不良事件,给药前后实验室检查未见异常值,表明在中国健康受试者中硝苯地平缓释片单次给药10 mg后的耐受性和安全性良好。
3 讨论
硝苯地平为第一个二氢吡啶类钙离子拮抗剂,因其疗效已得到充分肯定,且价格低廉,故在临床广泛用于高血压和心绞痛等的治疗,但其血药浓度极易出现波动,使得其在临床应用中的有效性和安全性存在个体差异。研究表明,制剂、种族、基因多态性、食物、合并用药等因素均会对硝苯地平口服制剂的药动学产生影响,进而可能导致降压效果的改变及不良反应的发生[6]。
虽然国内外已有不少关于液相色谱-质谱(LC-MS)法测定人血浆中硝苯地平浓度的方法学报道[7-9],但关于中国人群服用硝苯地平缓释片的药动学及生物等效性研究报道并不多见,且有限的报道均见于2016年我国开展仿制药质量和疗效一致性评价工作前,而既往研究设计中所选取的参比制剂均为当时上市的国产仿制制[10-13],非原研制剂,对现今工作的指导意义有限。如2011年WANG等[14]报道20例中国健康男性受试者空腹单次服用某国产硝苯地平缓释片10 mg药动学参数:Cmax(40.6±8.0)ng·mL-1、tmax(3.15±0.88)h、t1/2(7.53±2.10)h、AUC0-t(281.2±99.4)ng·h·mL-1、AUC0-∞(293.1±103.9)ng·h·mL-1,这与原研制剂Adalat®-L10说明书中公布6例日本健康受试者的部分药动学参数:Cmax(26.1±2.2)ng·mL-1和tmax(2.47±0.40)h存在一定差异[15]。
鉴于参比制剂在仿制药一致性评价中的关键作用,国家药品监督管理局药品评审中心于2016年起陆续发布仿制药参比制剂目录,并在第八批仿制药参比制剂目录中列出多种规格的硝苯地平缓释片、控释片、软胶囊的一致性评价工作均应选择拜耳公司Adalat系列产品作为参比制剂[16]。本项研究选择日本拜耳公司生产的10 mg硝苯地平缓释片(Adalat®-L10)作为参比制剂,结果显示受试制剂T1和参比制剂不等效,而改良后的受试制剂T2和参比制剂在空腹及餐后状态下生物等效。本研究也首次报道中国健康男性受试者空腹状态下单次口服Adalat®-L10的主要药动学参数, 结果显示Cmax、tmax等参数较既往报道[14]与Adalat®-L10说明书中日本健康受试者报道的数据更加接近[15],反映参比制剂选择的重要性。
此外,进食对不同剂型甚至同一剂型不同制剂的硝苯地平药动学可能产生不同的影响。CHALLENOR等[17]曾考察标准早餐(总能量约2800 J)对8例健康男性受试者单次服用10 mg硝苯地平胶囊的影响,结果显示饮食会显著延长tmax和降低Cmax(P<0.05),但对AUC0-t的降低差异无统计学意义。HIRASAWA等[18]考察标准早餐(总能量2790 J)对10例患者单次服用硝苯地平胶囊10 mg的影响,结果显示餐后状态下Cmax较空腹时显著降低(P<0.001),但tmax的延长和AUC0-t的降低差异无统计学意义。SCHUG等[19]通过四周期、四交叉试验考察高脂餐对24位健康男性受试者单次服用两种60 mg硝苯地平缓释片的影响,结果显示餐后状态下仿制制剂Nifedicron的药动学参数Cmax、AUC0-t、AUC0-t较空腹状态下显著提高(P<0.05),但原研制剂Adalat OROS的相关参数并未发生显著变化。在SCHUG开展的另一项纳入24位健康男性受试者的类似研究中,11位受试者在餐后状态下服用溶蚀骨架缓释制剂CORAL®的Cmax为空腹状态下的3~4倍,而缓释制剂Adalat OROS的药动学未发生显著变化[20]。
李江峰等[21]将90名健康受试者分为空腹组和餐后组,单次服用90 mg硝苯地平缓释片(未注明厂家)以考察高脂餐对其药动学的影响,结果显示餐后组tmax、Cmax和AUC0-t较空腹状态下分别升高31.9%、77.4%和30.0%(P<0.05),而t1/2、AUC0-∞仅略有升高,差异无统计学意义。本研究中,餐后组受试者单次口服Adalat®-L10的Cmax和tmax与空腹组差异无统计学意义,但AUC0-t、AUC0-∞和t1/2较空腹组均有显著降低。因本研究目的在于考察生物等效性而非饮食对硝苯地平缓释片药动学的影响,故试验设计上无法判断上述情况是否由受试者的个体差异造成。笔者认为,食物对硝苯地平缓释片生物利用度的影响较为复杂,未来有必要开展更多临床研究。
美国食品药品管理局(FDA)关于硝苯地平缓释片生物等效性指导原则中未说明该药物是高变异药物,本研究采用三交叉设计,受试制剂T1、T2分别与参比制剂比较。据既往研究的药动学数据,个体内变异≤20%。以药动学参数(AUC、Cmax)为主要分析指标,假定单侧α=0.05,β=0.1,Intra-CV=20%,受试制剂和参比制剂均值比值0.95,生物等效区间=80.00%~125.00%,采用SAS9.4软件计算,样本例数为26例。若以脱落率15%计,则空腹试验和餐后试验各入组30例受试者。
此外,体外溶出试验结果显示,3种制剂在不同pH值溶出介质中的释放行为相对稳定,但不同制剂间在溶解速率上差异有统计学意义。其中,受试制剂T1在4 h的释放率达到90%,而参比制剂和受试制剂T2在8~12 h后方达到90%的释放率。体外溶出实验结果显示,T1的体外释放比R和T2要快,与本文三制剂的体内生物等效性试验结果一致。
猜你喜欢
基层中医药(2022年7期)2022-11-17
健康体检与管理(2022年4期)2022-05-13
中国典型病例大全(2022年13期)2022-05-10
医学食疗与健康(2022年2期)2022-04-23
中国药学药品知识仓库(2022年5期)2022-04-11
中国药学药品知识仓库(2022年1期)2022-03-23
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
智慧健康(2021年33期)2021-03-16
恋爱婚姻家庭(2018年33期)2018-07-22
中国中药杂志(2016年23期)2017-04-07
