
TYMP基因新突变致孪生姐妹线粒体神经胃肠型脑肌病2例☆
2022-02-03 02:53:58窦炜康朱婷鸽赵旭霞兀瑞周慧敏史明
中国神经精神疾病杂志 2022年10期
窦炜康 朱婷鸽 赵旭霞 兀瑞 周慧敏 史明
线粒体神经胃肠型脑肌病(mitochondrial neurogastrointestinal encephalomyopathy, MNGIE)是一种极其罕见的常染色体隐性遗传疾病,已被证实由位于染色体22q13.32的TYMP基因突变引起[1]。该基因编码胸腺嘧啶磷酸化酶(thymidine phosphorylase, TP),突变后引起的酶缺乏会导致脱氧胸苷(deoxythymidine,dThd)和脱氧尿苷(deoxyuridine,dUrd)的全身性积累,从而导致相应的脱氧核糖核苷三磷酸盐(deoxy-ribonucleoside triphosphate,dNTP)在胞内浓度升高,影响线粒体DNA复制,最终导致线粒体DNA突变和功能异常[2]。患者常出现多系统受累症状,如胃肠道功能异常、恶病质、上睑下垂或进行性眼外肌麻痹、周围神经病变、弥漫性脑白质营养不良等。上述症状进行性恶化,常导致患者于成年早期死亡,平均死亡年龄为37.6岁[3]。本文报告2例患MNGIE的同卵双生姐妹,基因检测发现2例患者TYMP基因均存在两处新发错义突变,而其父母分别携带其中一处突变而未发病。本研究结果扩展了TYMP基因的遗传谱系,并对进一步认识MNGIE遗传发病机制有一定的借鉴。
1 临床资料
先证者,女,19岁,学生,因“四肢无力伴麻木1个月”于2021年1月入住我院神经内科。患者1个月前无明显诱因,逐渐出现四肢末端袜套感及脚踩棉花感,双手无法进行精细动作,尚可自主行走。曾就诊于当地医院,肌电图检查提示:四肢多发运动和感觉神经损害,其中感觉神经损害较重,考虑“吉兰-巴雷综合征”,接受醋酸泼尼松、维生素B1、维生素B12、神经节苷脂等营养神经治疗后,自觉症状有所减轻。为明确诊断和进一步治疗以“周围神经病”收住我院神经内科。发病以来,患者一般情况及精神、食欲欠佳,近期体质量明显减轻约3 kg。既往史:患者平素体健,近1年来反复查验肝功能异常,否认有肝炎、结核等病史。内科查体:体型偏瘦,贫血面容,身高约160 cm,体质量44 kg,BMI 17.19 kg/m2;心肺等查体无明显异常。专科查体:定向力、计算力、记忆力等高级神经功能正常,简易精神状态量表(mini-mental state examination,MMSE)评分28分;脑神经检查未见异常;上肢近端肌力5级,上肢远端、下肢肌力4级;双手手腕以下,双侧小腿下1/3以下痛温觉减退,双侧膝关节音叉震动觉减退、关节位置觉及图形复合觉减退;双侧腱反射均未引出,腹壁反射存在;各项病理征阴性;Kernig征和Brudzinski征阴性。家族史:父母身体健康,非近亲结婚,患者有一同卵双生姐姐,有类似临床表现,但症状相对较轻。
实验室及相关辅助检查:血常规血红蛋白 105 g/L;ALT 83 U/L,AST 74 U/L;血脂,TG 4.44 mmol/L;血沉28.0 mm/h;血浆乳酸,活动前4.97 mmol/L,活动后15 min 6.90 mmol/L。尿常规、甲状腺功能、凝血系列、血糖、心肌酶谱、性腺五项、生长激素大致正常,自身抗体谱及周围神经抗体(GM1、GM2、GD1a、GD1b等)阴性。脑脊液检查:白细胞计数40×106/L,蛋白990.0 mg/L,葡萄糖、氯化物正常;蛋白定量发现,白蛋白和免疫球蛋白(IgG、IgA、IgM等)均增高,QA1b中度升高,提示血脑屏障中度受损。骨髓穿刺:骨髓、粒系、红系增生活跃(考虑缺铁性贫血)。腹部超声:肝脏肿大,脾脏大小正常。头颅MRI可见广泛对称性脑白质病变(图1A-C)。神经电生理:四肢对称性周围神经损伤,运动和感觉神经均受累,轴索和髓鞘均有不同程度受损;肌电图显示肌源性损害。24 h视频脑电图:背景活动正常,睡眠分期清晰,监测全程未见异常波发放。肌肉活检病理报告(外院):改良Gomori染色可见2条破碎红纤维(ragged red fibers,RRFs);NADH染色部分肌纤维肌膜下深染;琥珀酸脱氢酶(succinate dehydrogenase,SDH)染色可见2条破碎蓝纤维(ragged blue fibers,RBFs)和2条SDH高反应性血管(SSVs);COX染色可见细胞色素氧化酶活性缺失肌纤维、PAS染色可见部分肌纤维内糖原含量减少、油红O染色可见部分肌纤维内脂滴含量增多。
考虑到患者的同卵双生姐姐亦有类似症状,征得同意后对姐姐进行了肝肾功能、腹部超声、头颅MRI、肌电图和脑电图等检查,发现姐姐同样存在转氨酶升高、肝脏肿大、头颅MRI显示广泛对称性脑白质病变(图1D-F)、肌电图提示多发性运动和感觉神经损害(下肢较上肢重,髓鞘损害较轴索重)和肌源性损害、24 h视频脑电图未见明显异常等表现。为了明确诊断,在患者及家属签署知情同意书后,采集孪生姐妹和其父母外周静脉血5 mL,提取基因组DNA行全基因组外显子测序(北京金准基因),并对可疑致病基因变异进行Sanger测序验证。结果发现孪生姐妹TYMP基因存在两处错义突变:3号外显子区域的c.239_263 delTTCGGGGCATGGATCTGGAGGAGAC(p.L80Pfs*4)和 7号外显子区域的c.776G>A(p.G259E)。第一处为缺失突变,导致氨基酸移码突变,使80号亮氨酸(L)突变为脯氨酸(P),并终止于下游4号密码子处;第二处为点突变,导致259号甘氨酸(G)突变为谷氨酸(E)。母亲仅存在第一处缺失突变,而父亲仅存在第二处突变。Sanger测序验证了上述突变(图2)。经检索HGMDpro数据库,发现上述两处变异均为未报道的新发突变。根据ACMG指南和标准,c.239_263 del和c.776G>A评级分别为“可能致病”和“临床意义未明”。

图1 孪生姐妹患者的头颅MRI影像 先证者(A-C)和其孪生姐姐(D-F)头颅MRI的T2-FLAIR序列示脑干、颞极、额叶、脑室周围、外囊及半卵圆中心等处广泛对称性脑白质病变。
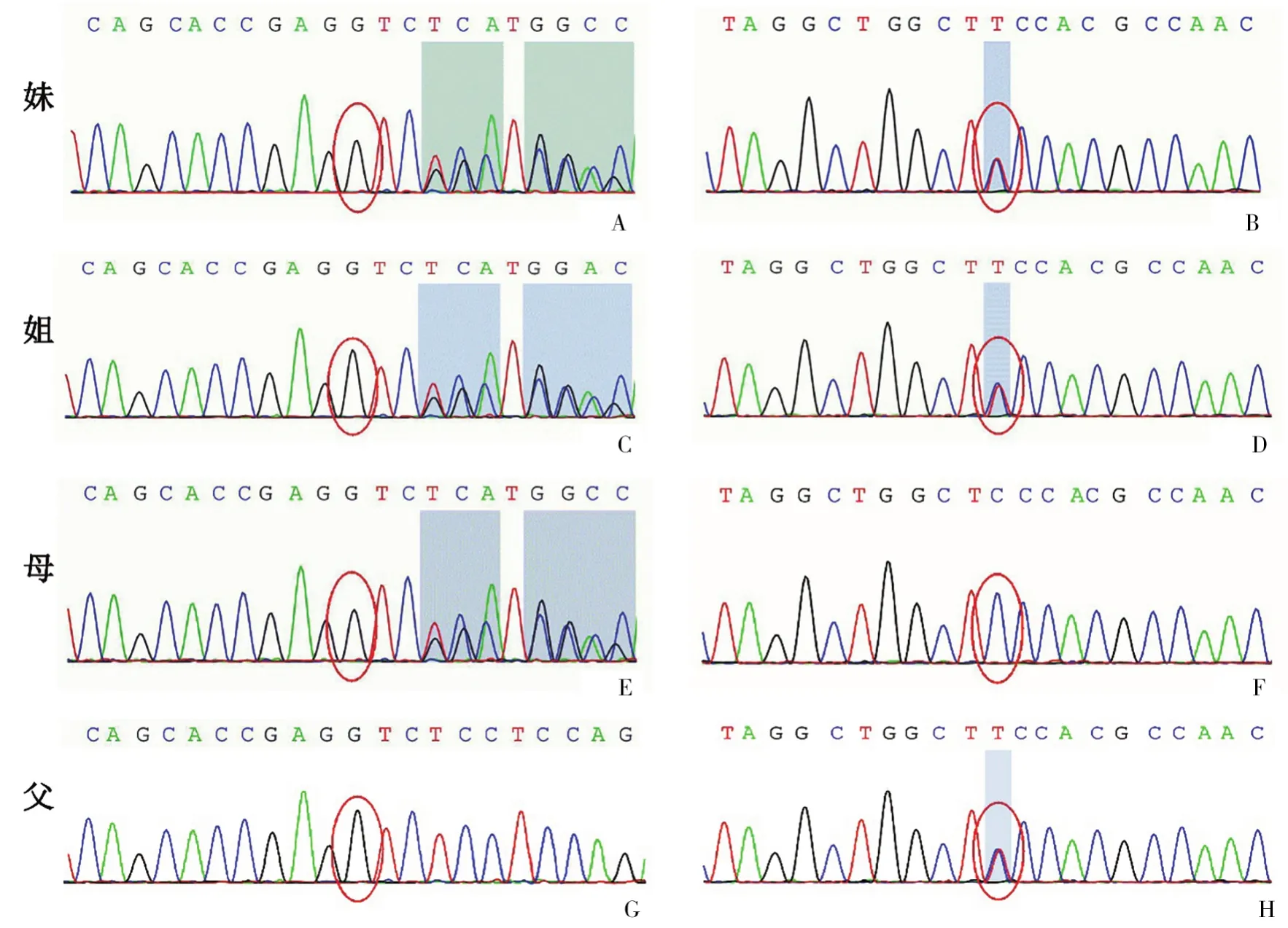
图2 孪生姐妹及其母亲TYMP基因的Sanger测序结果(均为反义链) 先证者(A)、孪生姐姐(C)和其母亲(E)均存在c.239_263 delTTCGGGGCATGGATCTGGAGGAGAC(p.L80Pfs*4)杂合变异,而父亲未发现此变异(G)。孪生姐妹(B, D)和其父亲(H)存在c.776G>A(p.G259E)杂合突变,但其母亲(F)未发现此变异。
根据孪生姐妹患者临床表现、辅助检查及基因测序结果,诊断为“线粒体神经胃肠型脑肌病”,因此停用激素,给予甲钴胺、复合维生素B、辅酶Q10、艾地苯醌等营养神经、改善线粒体功能治疗。患者诉四肢无力及麻木症状较前好转。出院时嘱患者坚持上述治疗,同时预防感染,避免极冷极热环境、过度锻炼、服用影响线粒体功能药物。1年后随访,患者体型仍消瘦、食欲差、易疲劳、转氨酶高,但四肢无力及麻木等症状缓解。
2 讨论
MNGIE是一种进行性的累及多系统的遗传性线粒体疾病,HIRANO等[4]于1994年首次对该病进行命名,该病的发病年龄根据报道可从5个月到50岁不等,平均发病年龄18.5岁[5-7]。关于其诊断当前国际上多采用HIRANO等[4]提出的标准:①上睑下垂或进行性眼外肌瘫痪;②严重的胃肠动力障碍;③表现为感觉异常和轻度肢体无力的周围神经病变,下肢较上肢更为严重;④具有线粒体肌病组织学特征的肌肉活检,例如可发现RRFs和SSVs。随着检测技术的发展,基因检测证实存在TYMP基因突变、头颅MRI显示弥漫性脑白质营养不良、检测白细胞TP活性、检测血浆或尿中的dThd、dUrd水平等也为MNGIE诊断提供了有力的证据。
在临床实际工作中,由于MNGIE的罕见性及复杂的多系统受累表现,患者通常要经过多年、转诊于不同科室才能获得正确的诊断,有报道诊断延误时间平均在5~10年[8],往往被误诊为克罗恩病、神经性厌食症、慢性假性肠梗阻、Whipple病、吉兰-巴雷综合征等[9]。由于疾病会对机体造成进行性损害,诊断延误会导致患者治疗反应性不佳和预后差。本例先证者以周围神经病起病,脑脊液检查发现蛋白显著增高,因此在当地诊断为“吉兰-巴雷综合征”,并给予激素治疗。但进一步头颅MRI检查发现弥漫性脑白质病变、肌肉活检发现典型的RRFs和SSVs、基因检测发现先证者和其孪生姐姐的TYMP基因有两处新发错义突变(c.239_263 del和c.776G>A),虽然此两处变异评级并不是“致病变异”,也缺乏典型的眼外肌瘫痪和严重胃肠道疾病等临床表现,但仍强烈提示MNGIE的诊断,因此对治疗方案进行了调整。
基因检测发现TYMP基因突变已经成为诊断MNGIE有力手段。截至2014年人类基因突变数据库已报告TYMP基因有97种不同的变异,包括错义、复制、删除、单碱基插入和内含子剪接等,涵盖外显子和内含子区域[10]。但新的TYMP基因突变位点仍被陆续发现,如董明明等[11]报告3号外显子c.417+1G>A纯合突变、周亚光等[12]报告7号外显子c.914T>C和10号外显子c.1319T>C两处错义突变可能是导致MNGIE的新发突变。本例中,全基因组外显子测序发现孪生姐妹患者的TYMP基因均存在两处杂合突变:c.239_263 del和c.776G>A,从而导致相应氨基酸发生突变。经查阅以往国内外文献并检索HGMDpro数据库,发现这两处变异为尚未报告的新发突变。根据ACMG指南,此两处突变评级分别为“可能致病”和“临床意义未明”。因患者母亲仅携带c.239_263 del,父亲仅携带c.776G>A突变,但均未发病。考虑到MNGIE是一种常染色体隐性遗传疾病。因此推测,该孪生姐妹TYMP等位基因同时存在母源性c.239_263 del和父源性c.776G>A突变从而导致MNGIE。然而,此两处变异是否为导致MNGIE的致病突变,还需在细胞学和动物学实验中进一步验证。
目前MNGIE尚缺乏特异治疗方法,以对症支持治疗为主,如给予辅酶Q10、B族维生素、维生素C、维生素E等药物以改善线粒体的功能,同时建议避免过度运动、极冷极热、防止感染,避免服用苯妥英钠、丙戊酸盐、氯霉素、利奈唑胺、氨基糖苷类抗生素及四环素等影响线粒体功能的药物[5]。此外一些旨在恢复患者生化平衡(如血浆中过高的dThd和dUrd水平)方法也在积极尝试中[13],如血液透析、持续非卧床腹膜透析、红细胞包裹TP输注和血小板输注等,可暂时有效改善一些MNGIE患者的生化失衡以缓解症状。然而,这些方法往往受制于患者本身或临床实际,在一定程度上应用受限,同时它们的安全性和长期临床效果亦有待进一步验证[14-16]。异基因造血干细胞移植[17]和原位肝移植[18]可永久性恢复TP功能,从而长期清除dUrd和dThd,以达到改善症状和稳定病情进展的目的,但受到供体限制,需要长期免疫抑制治疗,且与治疗相关的并发症和死亡率的风险很高[19-20]。
综上所述,本例报道以周围神经病发病的孪生姐妹患者,经全基因组外显子测序发现TYMP基因存在两处新发杂合突变:母源性c.239_263 del和父源性c.776G>A,结合辅助检查拟诊为MNGIE。由于MNGIE是一种罕见的常染色体隐性遗传病,临床表现多样、异质性强。如果对于以周围神经病变起病,伴有乳酸水平增高、弥漫性脑白质病变的患者,应考虑到此病的可能性。同时建议行基因检测以尽早明确诊断并给与规范化治疗,为患者提供最佳获益的机会。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29 01:57:52
海洋通报(2021年1期)2021-07-23 01:55:14
生物学通报(2021年4期)2021-03-16 05:41:26
中国生殖健康(2020年4期)2021-01-18 02:58:10
中国生殖健康(2018年4期)2018-11-06 07:12:16
河北医学(2016年5期)2016-12-01 03:58:56
实用临床医学(2016年8期)2016-06-07 01:28:23
中华老年多器官疾病杂志(2016年8期)2016-05-14 07:17:02
湖北农业科学(2014年11期)2014-09-10 18:06:07
癌变·畸变·突变(2014年1期)2014-03-01 04:39:36
