
固相萃取-高效液相色谱-串联质谱法测定蜂胶中氯霉素残留
2022-01-24 11:40王玉涵王欣然陆超丽吴殿军周金慧李熠
食品研究与开发 2022年1期
王玉涵,王欣然,陆超丽,吴殿军,周金慧,3,4*,李熠
(1.中国农业科学院蜜蜂研究所,北京 100093;2.昭平县农村社会事业促进站,广西 贺州 546800;3.农业农村部蜂产品质量安全控制重点实验室,北京 100093;4.农业农村部蜂产品质量安全风险评估实验室,北京 100093;5.中国农业科学院农产品加工研究所,北京 100193)
蜂胶,是蜜蜂采集胶源植物(杨树、桦树、桉树、酒神菊类植物、血桐属植物、黄檀属植物)树脂等分泌物后与其上颚腺、蜡腺等分泌物混合形成的胶黏性物质,被蜜蜂用来修补蜂箱的缝隙、加固以及清洁蜂巢[1-2]。蜂胶组成复杂,主要包括树脂(55%)、蜂蜡(30%)、挥发性油(10%)以及蜂花粉(5%)等组分,已知成分达420余种,其中包含多达70余种的黄酮化合物以及其他活性物质,丰富的活性成分赋予了蜂胶抗氧化、抗炎、抗菌和抗病毒等生物活性,使其具有增强机体免疫力和预防疾病等功效[3-5]。蜜蜂在生存环境条件恶劣以及自身免疫力较低时,容易受到病菌的侵害,导致蜂群感染欧洲幼虫腐臭病等细菌性疾病,蜂农常用抗菌性药物来防治这类蜜蜂疾病,其中包括具有广谱抗菌性且价格低廉的氯霉素[6]。然而药物的不规范使用会导致药物在蜂群中残留进而污染蜂产品。已有相关研究表明氯霉素可导致再生障碍性贫血和灰婴综合征[7],但目前尚无明确的剂量-效应作用[8]。目前很多国家已禁止在食用性动物中使用氯霉素药物,许多国家均规定氯霉素在动物源性食品中不得检出[9-11]。然而蜂胶中氯霉素残留检测的相关标准尚不完善,为保障蜂胶产品的质量安全,需建立对蜂胶中氯霉素药物残留可靠有效的检测方法。
目前为止,畜禽肉类、水产品、牛奶以及蜂蜜[8,12-14]等食品中氯霉素残留检测方法已多见报道,氯霉素的检测手段主要包括酶联免疫吸附法(enzyme-linked immunosorbent assay,ELISA)[15]、高效液相色谱法(high performance liquid chromatography,HPLC)[16]、气相色谱-质谱法(gas chromatography-mass spectrometry,GC-MS)[17]以及高效液相色谱-串联质谱法(high performance liquid chromatography-tandem mass spectrometry,HPLCMS/MS)[18],还包括酶生物传感器[19]和钨酸锌纳米电化学传感器[20]等新型检测方法。其中HPLC-MS/MS具有高灵敏度和稳定性,常与液-液萃取[21]和基体分散固相萃取(matrix solid-phase dispersion,MSPD)[14]等技术相结合,被广泛应用于食品中药物残留检测,实现对目标分析物的准确定量[22]。目前,对蜂胶中氯霉素提取多采用液-液萃取法,使用大量的对环境和人体健康不利的有机试剂如乙酸铅、甲基叔丁基醚、正己烷等[5,18,21,23]。此外,此类研究定量线性范围有限,未涵盖50 μg/L以上的较高浓度范围。因此,建立一种快捷、高效、绿色环保,同时可实现蜂胶中不同浓度氯霉素残留的准确定量的检测方法是十分必要的。
本研究通过HPLC-MS/MS法进行定性、定量分析测定蜂胶中氯霉素药物残留。考察不同提取溶剂、固相萃取柱等因素对蜂胶中残留氯霉素的提取、净化效率的影响。并对色谱和质谱条件进行优化,达到分析目标物所需的灵敏度和稳定性。因此本研究可为监控蜂胶质量安全提供技术支撑。
1 材料与方法
1.1 材料与试剂
蜂胶样品:市售;氯霉素(纯度98%)、氯霉素-D5(纯度98%):德国Dr.Ehrenstorfer公司;甲醇(色谱纯)、乙腈(色谱纯):美国Fisher公司;氯化钠、氢氧化钙(均为分析纯):上海阿拉丁生化科技股份有限公司;Bond Elut Plexa 固相萃取柱(200 mg,6 mL):美国 Agilent公司。
1.2 仪器与设备
Agilent 1290 HPLC高效液相色谱仪、6495三重四极杆串联质谱仪(配电喷雾电离源):美国Agilent公司;G560E涡动仪:美国Scientific Industries公司;1-15PK离心机:美国Sigma有限公司;Milli-Q纯水发生器:美国Millipore公司;BSA124S电子天平:德国赛多利斯有限公司;Kinetex C18色谱柱(50 mm×2.1 mm,2.6 μm):美国 Phenomenex公司。
1.3 标准溶液制备
准确称取氯霉素标准品及其内标氯霉素-D5标准品各10 mg,用甲醇溶解配制成1 000 mg/L的标准储备液,于-20℃棕色瓶中避光保存;用乙腈将氯霉素-D5标准储备液稀释为1 mg/L的标准使用液;将氯霉素标准储备液稀释制备浓度为 0.1、0.5、1.0、5.0、10.0、50.0、100.0、250.0、500.0 μg/L,内标质量浓度为 50 μg/L 系列标准工作溶液,供高效液相色谱-串联质谱仪测定。
1.4 样品前处理
1.4.1 样品制备
在温度高于25℃的条件下,蜂胶呈黏性胶质状态,将样品置于-20℃条件下冷冻2 h,取出冷冻样品立即用粉碎机制备蜂胶均匀粉末。
1.4.2 提取
称取蜂胶粉末0.25 g于50 mL离心管中,加入25 μL浓度为100 ng/mL的氯霉素-D5标准溶液,涡旋混匀。加入5 mL乙腈,涡旋至蜂胶完全溶解,加入5 mL 0.5%Ca(OH)2溶液,涡旋混匀,再加入 1 g NaCl,漩涡振荡2 min,于4℃下8 000 r/min离心10 min,取出上层清液,在40℃下氮吹至近干,加入10 mL 10%甲醇复溶,4℃下8 000 r/min离心10 min,保留上层清液,下层沉淀中加入10 mL 10%甲醇溶解,重复提取一次,将两次上清液合并,待固相萃取柱净化。
1.4.3 净化
Bond Elut Plexa固相萃取柱(6 mL,200 mg)使用前依次用10 mL甲醇和10 mL水活化,将1.4.2节中上清液转移至已活化的Bond Elut Plexa固相萃取柱中,调节流速以≤3 mL/min通过固相萃取柱。待上清液完全流出后,10 mL水淋洗,弃去流出液,负压抽干。10 mL甲醇洗脱,洗脱液收集于15 mL离心管中,于40℃下氮吹至近干,1 mL 10%乙腈水复溶,4℃下14 000 r/min离心10 min,上清液经0.22 μm滤膜过滤后进行HPLC-MS/MS测定。
1.5 色谱条件
流动相 A:水;流动相 B:乙腈。梯度洗脱:0~2 min,10%B;2 min~4 min,30%B;4 min~4.1 min,75%B;4.1 min~5.1 min,100%B;5.1 min~6.5 min 10%B。流速:0.4 mL/min;柱温:40 ℃;进样量:5 μL。
1.6 质谱条件
电喷雾离子源(electrospray ionization source,ESI);负离子模式;扫描方式:多反应监测模式(multiple reaction monitoring,MRM);毛细管电压:2 500 V;干燥气温度:270℃;干燥气流速:11L/min;雾化器压力:275.8kPa;鞘气温度:350℃;鞘气流速:12 L/min;喷嘴电压:0 V。
2 结果与讨论
2.1 质谱条件
氯霉素在负离子模式下可获得稳定的分子离子[M-H]-。本试验中分别将1.0 mg/L的氯霉素标准溶液和1.0 mg/L的氯霉素-D5标准溶液注入电喷雾离子源,在负离子(ESI-)全扫描模式下获得氯霉素和氯霉素-D5的分子离子[M-H]-。然后在碎片离子扫描模式下得到氯霉素和氯霉素-D5的子离子信息。最后通过多反应监测(MRM)扫描模式对各子离子碰撞能进行优化,获得氯霉素和氯霉素-D5的最佳质谱参数,质谱选择离子参数见表1。

表1 质谱选择离子参数Table 1 Transitions and optimal conditions used for MS/MS analysis
2.2 色谱条件确定
本研究考察了甲醇/水和乙腈/水两种不同的流动相体系,由于乙腈的极性比甲醇弱,它更能有效地洗脱弱极性的氯霉素,缩短氯霉素在色谱柱中的保留时间,同时在乙腈/水体系中干扰峰少,且氯霉素获得了较高的质谱响应。通过调整流动相洗脱比例,可使目标分析物尽可能远离杂质干扰峰产生的离子抑制区域,以降低基质影响并提高目标分析物的质谱响应[24-25]。本研究中发现当乙腈比例快速增加时,可缩短目标分析物的保留时间并获得更高的信号响应。色谱柱的温度也会影响分析时间和目标物的分离效果,为了在合适的分析时间内完成目标物的有效分离,对不同柱温(30、35、40℃)进行了测试和比较。结果表明,目标分析物在柱温为40℃时分离效果最佳。因此,本研究中采用乙腈/水作为流动相,40℃柱温,经优化的色谱柱流动相梯度洗脱,在6.5 min(含清洗和平衡时间)的分离时间内,可实现良好的色谱分离并获得较高的质谱响应。
经优化的质谱和液相色谱条件下空白蜂胶基质、加标基质(2.0 μg/kg)以及氯霉素标准溶液(0.5 μg/L)的MRM色谱图见图1。

图1 MRM色谱图Fig.1 MRM chromatograms
2.3 样品前处理方法优化
2.3.1 提取条件优化
氯霉素的油水分配系数对数值为1.02,是偏亲脂性物质,可溶于有机溶剂,微溶于水。本研究中考察了甲醇、乙酸乙酯、乙腈以及甲苯/水溶液对蜂胶中氯霉素的提取效果,结果如图2所示。

图2 不同提取溶剂提取蜂胶中氯霉素的绝对回收率Fig.2 Absolute recoveries of chloramphenicol inpropolis using different extraction solutions
由图2可知,乙酸乙酯作提取剂时氯霉素的绝对回收率最高,但乙酸乙酯作提取溶剂时提取溶液浑浊,由于蜂胶中含有较多的脂溶性成分,因此在乙酸乙酯共同提取物中存在了较多脂溶性杂质(树脂、蜂蜡以及黄酮类化合物[26],这些共同提取物产生的杂质干扰峰影响目标物在质谱中的响应,不利于后续的分析测定。以甲醇∶水(体积比3∶7)作提取剂时,氯霉素绝对回收率为38%;当乙腈作为提取剂时,绝对回收率为59%,乙腈提取物相比于乙酸乙酯提取物澄清,且乙腈提取物的色谱图中杂质峰与目标分析物分离度高,基质干扰较小,利于低浓度目标物的筛查测定。当甲苯/水体系作为提取剂时,由于蜂胶可溶于甲苯,而氯霉素不溶于甲苯但可微溶于水,使得蜂胶基质中的脂溶性物质萃取到甲苯层而氯霉素萃取到水层,进而实现氯霉素与蜂胶基质中亲脂性杂质干扰物的分离。同时,不同比例的甲苯/水体系可导致提取效果有所差异,本研究测试了不同比例的甲苯∶水(体积比 7∶3、6∶4、5∶5、4∶6)的提取效果,其中甲苯∶水体积比为 6∶4 时可获得较高的绝对回收率(51.68%);甲苯∶水体积比 5∶5、4∶6时绝对回收率均近似为45%;甲苯∶水体积比为7∶3时,甲苯/水的萃取回收率最低,仅为41.57%。同时发现随着甲苯比例的增加,萃取过程发生了乳化,因此过高的甲苯比例不利于目标分析物的萃取分离。由以上结果表明,乙腈和甲苯∶水(体积比6∶4)作为提取溶剂时氯霉素的绝对回收率较高,同时共同提取物干扰少,有利于后续分析测定。由于甲苯的毒性较大且使用时危险性较高,因此,最终选用乙腈作为提取剂。
为除去乙腈提取物中的杂质干扰,需对提取物进行除杂处理。本研究中分别测试了不同浓度的碱性溶液[Ca(OH)2溶液、NaOH 溶液]、酸性溶液(三氯乙酸)以及酸碱复合溶液的除杂效果,结果如图3所示。

图3 不同除杂溶液作用下蜂胶中氯霉素绝对回收率Fig.3 Absolute recoveries of chloramphenicol inpropolis using different solutions to remove impurities
由图3可知,采用1%NaOH溶液联合5%三氯乙酸除杂所获得绝对回收率最差(3%),由于黄酮类化合物可溶于碱性溶液,同时在酸性溶液中沉淀析出[27],在乙腈提取物中先后加入1%NaOH溶液和5%三氯乙酸溶液,出现了大量悬浮的絮状物,由于絮状物可能包裹目标分析物导致回收率变差;5%三氯乙酸除杂所获得绝对回收率为20%,由于氯霉素在酸性环境中易形成离子态,使得目标分析物的极性增大,难以被有机溶剂提取;1%NaOH溶液绝对回收率(58%)与0.5%Ca(OH)2溶液接近,当NaOH溶液与乙腈提取物混合时,同样出现明显絮凝现象,同时絮凝物与液体分层不明显。浓度为0.5%的Ca(OH)2溶液绝对回收率最高(66.8%),且絮凝物在静置3 min后可聚集并下沉到下层。本研究结果与杨黎等[28]的研究结果一致,0.5%Ca(OH)2溶液可用于去除乙腈蜂胶提取物中的亲脂性物质。
为促进液液萃取时杂质萃取层与目标分析物萃取层的分离,参考Koltsakidou等[29]的研究,在乙腈提取物与0.5%Ca(OH)2溶液混合体系中加入NaCl以促进两相分层。最后,在固相萃取柱净化前,需将样品溶液的溶剂体系转化为高比例水相体系,由于10%甲醇可以溶解氯霉素,同时不会破坏固相萃取柱的填料结构,因此,本研究采用10%甲醇对氮吹后的浓缩提取物进行复溶。
2.3.2 固相萃取小柱的选择及净化条件优化
经纯化的乙腈提取物呈淡黄色,表明其中黄酮类化合物未被完全除去。为进一步净化和保留富集乙腈提取物中的氯霉素,本研究比较了混合型阳离子交换固相萃取柱(PCX)、亲水亲脂型反相固相萃取柱(Oasis HLB)以及非极性保留固相萃取柱(Bond Elut Plexa)的净化效果。PCX和Oasis HLB固相萃取柱对添加氯霉素浓度水平为40 μg/kg的蜂胶基质进行富集净化,Bond Elut Plexa对添加氯霉素浓度水平为4 μg/kg的蜂胶基质进行富集净化,添加氯霉素的蜂胶基质经不同固相萃取柱净化后获得MRM色谱图如图4所示。

图4 不同固相萃取柱净化蜂胶中氯霉素的MRM色谱图Fig.4 MRM chromatograms of residual chloramphenicol in propolis under different solid-phase extraction cartridges
由图4可知,Oasis HLB柱相比于PCX柱,其对目标物的净化效果更好,可获得较高的目标物质谱信号响应,但其对添加浓度水平为4 μg/kg的提取物的净化富集效果不佳。为净化低添加浓度水平的提取物,本研究中考察了Bond Elut Plexa柱的净化能力,由于Bond Elut Plexa柱是以表面含羟基配体的亲水苯乙烯-二乙烯苯聚合物为填料[30],因此,能够保留具有羟基结构的氯霉素同时还能去除脂类杂质。由图4中Bond Elut Plexa固相萃取柱净化加标提取物中氯霉素的MRM色谱图可知,Bond Elut Plexa对基质净化效果明显,降低了杂质干扰并获得了较高的信号响应。在整个提取、除杂以及净化过程中都使用了与氯霉素结构和化学性质相似的同位素内标物氯霉素-D5,以校正分析物在前处理过程中的质量损失,在最佳提取和净化条件下,获得了90%以上的相对回收率。
2.4 方法验证
2.4.1 方法线性关系
用乙腈溶剂配制浓度为 0.1、0.5、1.0、5.0、10.0、50.0、100.0、250.0、500.0 μg/L,内标物氯霉素-D5 浓度均为2.5 μg/L的系列标准工作液,在优化后的质谱和色谱条件下进行测定。以所对应的氯霉素标准溶液浓度为横坐标(X),各浓度下氯霉素色谱峰面积平均值为纵坐标(Y),外标法线性回归方程为Y=31 177X+105 316(R2=0.998 5);以氯霉素标准溶液浓度为横坐标(X),各浓度下氯霉素色谱峰面积与氯霉素-D5色谱峰面积比值的平均值为纵坐标(Y),内标法的线性回归方程为 Y=0.413 2X-1.237 9(R2=0.999 2)。综上,采用同位素内标校正,线性相关系数和回收率均优于外标法,因此,本研究采用内标法定量,氯霉素在0.1 μg/L~500.0 μg/L浓度范围内,其浓度与对应的其定量离子峰面积比值呈良好线性关系,线性相关系数大于0.99,满足残留分析的要求。
2.4.2 基质效应评价
由于基质中存在共提取物,会增强或抑制待测目标物的信号响应,影响分析结果的准确性。为对目标物进行准确定量,需对样品的基质效应进行评估。由1.4节前处理操作制备得到含内标物的空白基质液,在0.1 μg/L~500.0 μg/L浓度范围内配制与上述含内标物的系列标准工作液浓度相同的系列基质标准工作液,并计算出含内标物的样品基质匹配标准曲线方程Y=733.36X+2 166.8(R2=0.999 0)。基质效应计算公式为B/A×100(A为含内标物的溶剂标准校准曲线的斜率;B为含内标物的基质匹配标准曲线的斜率),当基质效应在80%到120%之间被认为是可被接受的低基质效应,小于80%被认为存在基质抑制效应;大于120%被判定为存在基质增强效应[31]。结果表明,经内标物校正的基质效应为85.16%,基质抑制效应低于20%,因此本试验中可采用含内标物的溶剂配制标准工作曲线溶液进行定量分析。
2.4.3 方法检出限
按2.4.2方法得到含内标物的空白基质液,添加系列浓度的氯霉素标准溶液并进行测定分析,根据6个空白样品中基线噪音的平均值,以3倍信噪比(S/N=3)计算得到该方法检测限;以10倍信噪比(S/N=10)计算得到该方法定量限。经测定本方法检测限(limit of detection,LOD)和定量限(limit of quantification,LOQ)分别为 0.4 μg/kg 和 1 μg/kg。
2.4.4 方法加标回收率和精密度
在含内标物的空白蜂胶样品中分别添加1、2、10 μg/kg 3个同浓度水平,每个水平设5个平行,设置一组空白对照。计算得到回收率及精密度,测定结果见表2。
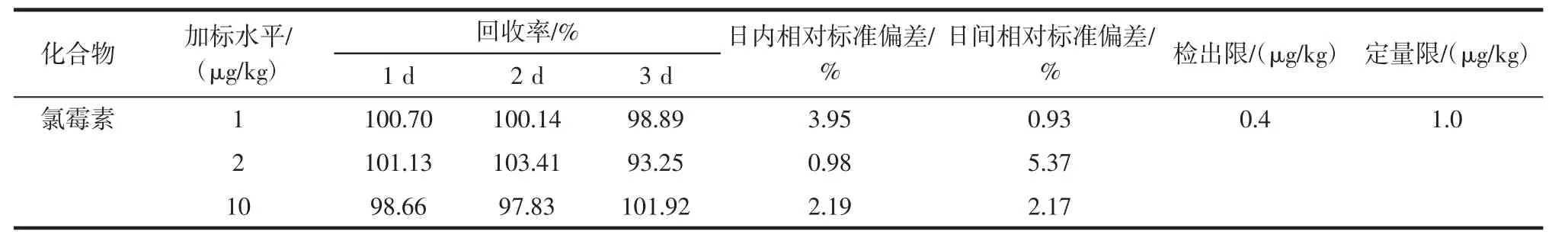
表2 方法回收率、精密度、检测限和定量限(n=5)Table 2 Recovery,precision,LOD and LOQ of CAP in blank propolis samples(n=5)
蜂胶样品中氯霉素在3个添加浓度的回收率在93.25%~103.41%之间。日内相对标准偏差在0.98%~3.95%;日间相对标准偏差在0.93%~5.37%。由回收率和精密度可见,该方法准确性、稳定性良好,可满足残留检测的要求。
2.5 实际样品测定
采用本文所建立的方法测定2019年不同蜂场、蜂产品公司和超市的26份蜂胶样品。测定结果显示,15.38%的蜂胶样品检出氯霉素,其浓度在1.06 μg/kg~130.62 μg/kg之间。表明目前我国养蜂业中存在违规使用氯霉素的情况,因此,需加强对养蜂业中氯霉素药物的监督管理。
3 结论
本研究建立了固相萃取结合高效液相色谱-串联质谱法测定蜂胶中氯霉素残留的定性筛查与定量分析方法。经优化的前处理方法便捷、高效,经乙腈提取,经Bond Elut Plexa固相萃取柱净化蜂胶基质,可实现蜂胶中氯霉素残留物的有效提取与富集净化。该方法的准确度、精密度和灵敏度均经过方法学评价,满足残留检测的要求,本方法可用于准确检测蜂胶中氯霉素残留水平。
猜你喜欢
食品安全导刊(2021年20期)2021-08-30
绿色科技(2021年12期)2021-07-22
化工自动化及仪表(2021年3期)2021-06-04
食品工程(2020年4期)2021-01-20
蜜蜂杂志(2020年5期)2020-12-02
酿酒科技(2019年10期)2019-11-12
中国蜂业(2019年3期)2019-04-03
渔业致富指南(2019年9期)2019-01-06
黄河黄土黄种人(2018年7期)2018-09-23
家庭科学·新健康(2016年5期)2016-05-12
