
巴洛沙韦关键中间体的合成工艺改进
2022-01-23 05:30:25于立国孙光祥张云然陶维洁
上海医药 2022年1期
于立国 孙光祥 张云然 陶维洁
摘 要 本研究报道了一条巴洛沙韦关键中间体3-(苄氧基)-4-氧代-4H-吡喃-2-羧酸(1)的新合成路线。以麦芽醇作为原料,经过苄基保护、缩合、氧化制得1。我们设计先将甲基间接转变成烯胺,再将烯氨氧化成醛,與甲基直接氧化成醛相比,避免使用剧毒品二氧化硒,消除了对环境的污染,并且有效提高路线的安全性。同时,本研究对烯胺的氧化条件进行了选择与优化,先将烯胺氧化成醛,醛中间体无需分离,进一步氧化到羧酸,“一锅法”实现了两个氧化历程,并且避免使用昂贵的钌催化剂,大幅度降低成本。总收率69.7%,产品纯度99.26%。
关键词 巴洛沙韦 产业化 合成
中图分类号:O625.8 文献标志码:A 文章编号:1006-1533(2022)01-0067-03
Improvement of synthetic process for key baloxavir intermediate
YU Liguo, SUN Guangxiang, ZHANG Yunran, TAO Weijie
(Changzhou Pharmaceutical Factory, Shanghai Pharmaceutical Group, Changzhou 213018, China)
ABSTRACT We reported a new synthetic route for baloxavir’s key intermediate 3-(benzyloxy)-4-oxo-4H-pyran-2-carboxylic acid (1). Maltol was used as the material and 1 was prepared by benzyl protection, condensation and oxidation. We designed the indirect conversion of methyl into enamine and then the oxidation of enamine into aldehyde. Compared with the direct oxidation of methyl into aldehyde, the new rout could avoid the use of selenium dioxide, eliminate the pollution to the environment and effectively improve the safety of the synthesis. Meanwhile, the oxidation conditions of enamine were selected and optimized in this study. The enamine was oxidized to aldehyde first, and the aldehyde intermediate was further oxidized to carboxylic acid without separation. The “one-pot method” realized two oxidation processes and avoided the use of expensive ruthenium catalyst, which could greatly reduce the cost. The total yield was 69.7% and the purity was 99.26%.
KEy wORDS baloxavir; industrialization; synthesis
巴洛沙韦(baloxavir,商品名Xofluza)是一种首创(first-in-class)、单剂量口服药物,具有不同于市面其他抗病毒药物的全新抗流感作用机制[1],是一种对流感病毒的复制必不可少的内切核酸酶抑制剂。巴洛沙韦具有非常好的市场潜力,与2009年销售额达32亿美元的竞争品种罗氏公司的奥司他韦相比[2],具有更快的抗病毒疗效,服药方案简单,不良反应更小,有望成为继奥司他韦后继续领跑抗流感药的王牌之一。
文献报道的3-(苄氧基)-4-氧代-4H-吡喃-2-羧酸(1)的合成路线主要有两条。①以麦芽醇为起始原料,经过苄基保护,制得3-(苄氧基)-2-甲基-4H-吡喃-4-酮(2),2经过二氧化硒氧化,制得3-(苄氧基)-4-氧基-4H-吡喃-2-甲醛(3),3经过亚氯酸钠氧化制得1[3]。其环境污染严重(须使用剧毒品二氧化硒[4]),反应温度高(155℃),收率较低,不适合工业化生产。②以麦芽醇为起始原料,经过苄基保护,制得2,2用LiHMDS拔氢后与苯甲醛发生缩合反应,制得3-(苄氧基)-2-(2-羟基-2-苯乙基)-4H-吡喃-4-酮(4),4与甲磺酰氯取代生成活性甲磺酸酯,在DBU存在的情况下发生脱水反应,生成(E)-3-(苄氧基)-2-苯乙烯基-4H-吡喃-4-酮(5),5在三氯化钌和高碘酸钠的条件下发生氧化反应,生成3,3经过亚氯酸钠氧化制得1[5]。其合成路线长,成本高(须使用钌催化剂[6]),加之环境污染严重,也不适合工业化生产。
1是合成巴洛沙韦的关键中间体,鉴于巴洛沙韦巨大的药用前景,开发一条环境友好、成本低、适合产业化的合成路线变得尤为重要。
1 材料与方法
1.1 仪器
LC-20AD高效液相色谱仪(日本岛津公司);ZF-8型暗箱式四用紫外线分析仪(上海嘉鹏科技有限公司);GF254薄层层析(TLC)板、P6107-C电子天平(阿拉丁公司)。
1.2 试剂
麦芽醇、N,N-二甲基甲酰胺二甲缩醛(DMF-DMA)及其他试剂均为市售工业级商品。
1.3 方法
1.3.1 关键中间体1合成路线
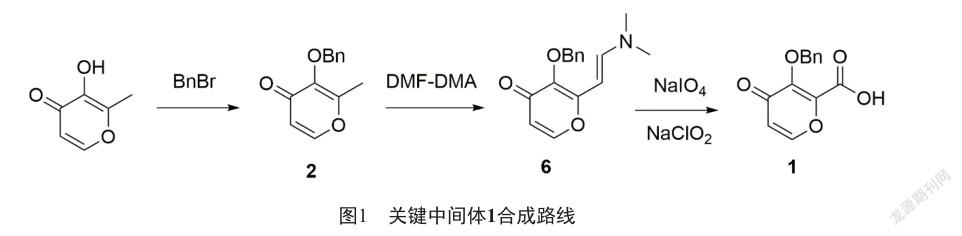
我们开发了一条环境友好、成本低、适合产业化的新合成路线(图1)。该路线以廉价易得的麦芽醇为起始原料,经过苄基保护,制得2,2与N,N-二甲基甲酰胺二甲基缩醛缩合生成(E)-3-(芐氧基)-2-(2-(二甲氨基)乙烯基)-4H-吡喃-4-酮(6),6经过高碘酸钠[7]和亚氯酸钠氧化[8]制得1。
1.3.2 HPLC检测
采用峰面积归一化法:Waters SunFire C18柱(4.6 mm×150 mm, 5mm);流动相 A 为0.01 mol/L Na2HPO4-三乙胺(TEA)=100∶0.1(H3PO4调pH=6.0)、B为甲醇,梯度洗脱(0~20 min:A 80%;20~25 min:A 50%;25~45 min:A 10%);柱温35 ℃;流速1.0 mL/min;检测波长225 nm;进样量10 mL;保留时间 22.6 min。
2 结果
2.1 化合物2的制备
将麦芽醇(1 890 g,14.99 mol)溶于18.9 L N,N-二甲基甲酰胺,加入溴化苄(1.84 L, 15.49 mol),碳酸钾(2 280 g,16.50 mol),加热到80 ℃,保温2 h,薄层色谱法检测原料完全转化,降温至室温,加入10 L四氢呋喃,抽滤,滤液于45 ℃减压浓缩至干得到白色固体(3 290 g,15.21 mol),收率100%。
1H NMR(400 MHz, CDCl3)δ 7.59(d,J=5.6 Hz, 1H)、7.42~7.29(m,5H)、6.37(d,J=5.6 Hz,1H)、5.16(s,2H)、2.08(s,3H)。
2.2 化合物6的制备
将化合物2(1 080 g,4.99 mol)溶于5.4 L N,N-二甲基甲酰胺,加入N,N-二甲基甲酰胺二甲基缩醛(1 480 g,12.42 mol),加热至回流,保温8 h,薄层色谱法检测原料完全转化,降温至室温,反应液中加入40 L水,16 L二氯甲烷萃取3次,合并有机层,弃去水层,有机层用适量无水硫酸钠干燥,抽滤,滤液于30 ℃减压浓缩至干,得到油状物(1 153 g,4.25 mol),收率85%。
1H NMR(400 MHz,CDCl3) δ 7.45(d,J=6.4 Hz,2H)、7.41(d,J=5.6 Hz,1H)、7.36~7.27(m,3H)、7.02(d,J=13.4 Hz,1H)、6.21(d,J=5.6 Hz,1H)、5.10(s,2H)、5.00(d,J=13.4 Hz,1H)、2.83(s,6H)。
2.3 化合物1的制备
将化合物6(1 466 g,5.40 mol)溶于7 L N,N-二甲基甲酰胺,加入高碘酸钠(2 310 g,10.80 mol)的15 L水溶液中,过程中控制反应液温度不超过25 ℃,加毕,反应液室温搅拌1 h,薄层色谱法检测原料完全转化,抽滤,滤饼用2 L乙酸乙酯漂洗,合并滤液,滤液用10 L乙酸乙酯萃取3次,合并有机层,有机层用5 L饱和氯化钠洗涤三次,有机层用无水硫酸钠干燥,抽滤,滤液浓缩得到油状物。将亚氯酸钠(1 466 g,16.21 mol)和氨基磺酸(1 052 g,10.84 mol)溶于7 L水中,将上述油状物用7 L丙酮溶解,滴加到水溶液中,控制温度不超过25 ℃,保温1 h,薄层色谱法检测原料完全转化,将反应液浓缩除去丙酮,析出大量白色固体,降温至5 ℃,抽滤,鼓风干燥,得到白色固体(1 092 g,4.44 mol),收率82%。HPLC检测产品纯度为99.26%。
1H NMR (400 MHz, DMSO) δ 14.23(s,1H)、8.19(d,J=5.6 Hz,1H)、7.42(d,J=6.7 Hz,2H)、7.36~7.29(m,3H)、6.54(d,J=5.6 Hz,1H)、5.10(s,2H)。
3 讨论
本工艺与现有文献方法相比,有如下优点:①先将甲基转变成烯胺,再氧化成醛,避免使用剧毒的二氧化硒,大幅减少环境污染;同时显著提高氧化的收率,有效降低了成本。②构建烯胺仅一步反应,与构建碳碳双键相比,既避免强碱低温给车间操作带来的麻烦,又避免使用剧毒品甲磺酰氯,绿色环保,大幅缩短反应步骤,收率很高,有利于成本控制。③通过对反应条件的优化,实现“一锅法”将烯胺氧化到羧酸,与碳碳双键的氧化相比,无需使用昂贵的三氯化钌,且反应收率更高。
改进后的工艺,反应条件温和、操作简便、手性纯度高、成本低廉,总收率69.7%,产品纯度99.26%,适合工业化生产。
致谢:感谢浙江大学为本研究的核磁共振检测提供支持。
参考文献
[1] 李佳悦, 刘洋. 巴洛沙韦[J] 中国药物化学杂志, 2019, 29(5): 411.
[2] 李秋, 王珊. 抗病毒药物的研究进展[J]. 医药导报, 2011, 30(6): 732-735.
[3] Puerta DT, Botta M, Jocher CJ, et al. Tris(pyrone) chelates of Gd(Ⅲ) as high solubility MRI-CA[J]. J Am Chem Soc, 2006, 128(7): 2222-2223.
[4] 蓝柳恒. 二氧化硒生产过程职业病危害的预防及控制[J].中国卫生产业, 2015, 12(11): 6-8.
[5] Aoyama Y, Hakogi T, Fukui Y, et al. Practical and scalable synthetic method for preparation of dolutegravir sodium: improvement of a synthetic route for large-scale synthesis[J]. Org Process Res Dev, 2019, 23(4): 565-570.
[6] 龙思, 宇裴响, 林罗丹, 等. 贵金属钌基催化剂合成及应用研究进展[J]. 化学通报, 2021, 84(2): 120-128.
[7] Bernd P. Selectivity versus reactivity-recent advances in RuO4-catalyzed oxidations[J]. Synthesis, 2005(15): 2453-2472.
[8] 姜志猛. 亚氯酸钠体系选择性氧化制备布洛芬及其它羧酸[J]. 中国医药工业杂志, 1991, 22(1): 1-2.
猜你喜欢
纺织科学研究(2021年9期)2021-10-14 08:52:08
今日农业(2019年14期)2019-09-18 01:21:42
宝藏(2018年12期)2019-01-29 01:51:34
中国化肥信息(2019年4期)2019-01-17 18:47:06
知识经济·中国直销(2017年7期)2017-07-24 14:12:38
现代商贸工业(2016年14期)2016-12-27 15:35:49
安徽理工大学学报·自然科学版(2016年4期)2016-12-23 09:55:25
考试周刊(2016年85期)2016-11-11 02:09:06
农业与技术(2016年15期)2016-11-09 07:10:15
科技视界(2016年18期)2016-11-03 00:34:31
