
超高效液相色谱-串联质谱法检测 蒸馏酒中的3种非法添加物质
2022-01-20 06:31:58王东江黎姝蔓
现代食品 2021年23期
◎ 杨 针,王东江,黎姝蔓,邓 雄
(重庆市永川食品药品检验所,重庆 402160)
我国白酒多以高粱、小麦等谷物为原料发酵后蒸馏储藏所制,以蒸馏酒为主导[1]。大多数白酒中含有一定比例的醛类、甲醇和杂醇油等物质,这些物质易引发饮酒者的上头、口干等不适反应。名优白酒往往通过改进酿造工艺提升酸类、酯类等香气物质含量,降低醛类等有害物质含量,然而,一些不法商贩违规在白酒中添加头痛粉(主要成分为对乙酰氨基酚、水杨酸和咖啡因)以缓解劣质白酒引起的头痛等不适症状,投机取巧,以次充好。头痛粉作为药品被添加入白酒中本就是非法添加成分[2],其药品说明及病例报道也明确表示其与酒精混服对肝功能有损伤[3-4]。
目前,针对头痛粉及其主要成分出现在白酒中的不良现象报道较多,其作为食品中的非法添加物检测采用是高效液相色谱法和气相色谱-串联质谱法[5-7],但检测方法存在灵敏度不高、分辨率不高、回收率不高、操作复杂、需不断更换流动相及分析时间长等不足,相关方法无法满足高灵敏度与较强抗干扰能力检测微量或痕量成分的需求。因此本研究旨在建立蒸馏酒中对乙酰氨基酚、水杨酸、咖啡因的UPLC-MS/MS检测方法,为蒸馏酒中非法添加头痛粉的准确、快速判定检测提供科学依据,为市场监管部门执法提供有力的技术支撑,从而达到保障人们饮食用药安全的 目的。
1 材料与方法
1.1 材料与试剂
水杨酸标准对照品,坛墨质检-标准物质中心,批号为1921804,含量为99.7%;对乙酰氨基酚标准对照品,坛墨质检-标准物质中心,批号为1201801,含量为98.0%;咖啡因标准对照品,坛墨质检-标准物质中心,批号为0981904,含量为98.0%;蒸馏酒酒样,市售;甲酸(色谱纯),山东西亚化学股份有限公司;甲醇(色谱纯),Merck KGaA。
1.2 仪器与设备
Agilent 1290型高效液相色谱仪(包括二元泵,在线脱气机,自动进样器),Agilent 6420型三重四极杆质谱仪(包括电喷雾离子源,Masshunter数据工作站),美国Agilent公司;XP205型万分之一电子天平,瑞士梅特勒-托利多仪器公司;JA5003N型千分之一电子天平,上海佑科仪器仪表有限公司;HH-8型水浴锅,江苏盛蓝仪器制造有限公司;Mtegral5型超纯水机,美国密理博公司。
1.3 方法
1.3.1 标准物质溶液的配制
标准储备液:分别精密称定对照品对乙酰氨基酚10.50 mg,咖啡因10.60 mg,水杨酸10.90 mg置于 25 mL容量瓶中,用甲醇溶解并定容至刻度,摇匀。分别得到对乙酰氨基酚储备液(411.60 μg·mL-1)、咖啡因储 备液(415.52 μg·mL-1)和水杨酸储备液 (434.69 μg·mL-1),-18 ℃低温避光保存。
混合标准中间液:分别精密移取对乙酰氨基酚、水杨酸和咖啡因储备液各2.5 mL于同一20 mL容量瓶中,用超纯水稀释定容至刻度并摇匀,得到混混合标准中间液,乙酰氨基酚、水杨酸和咖啡因的浓度分别为51.450 μg·mL-1、51.940 μg·mL-1和54.336 μg·mL-1, 4 ℃避光保存。
混合标准工作液:分别精密移取对乙酰氨基酚、水杨酸和咖啡因对照品储备液各0.1 mL于同一20 mL容量瓶中,用超纯水稀释定容至刻度并摇匀,得到混合标准工作液,乙酰氨基酚、水杨酸和咖啡因的浓度分别为2.058 μg·mL-1、2.078 μg·mL-1和2.173 μg·mL-1,4 ℃避光保存。
混合标准曲线系列工作液:分别吸取适量体积的混合 标准工作液,用水稀释,配制成浓度分别为0 ng·mL-1、 10 ng·mL-1、20 ng·mL-1、50 ng·mL-1、80 ng·mL-1、 100 ng·mL-1和120 ng·mL-1的系列标准工作溶液。
1.3.2 样品前处理优化
准确称取5 g试样(精确至0.01 g)置于50 mL烧杯中,于60 ℃水浴加热30 min,残渣全部转移至50 mL容量瓶中,用纯水定容至刻度并摇匀,经0.22 μm水相微孔滤膜过滤,供液相色谱-串联质谱分析。
1.3.3 色谱条件优化
色谱柱:ZORBAX RRHD SB-C18(2.1 mm×100 mm, 1.8 μm);流速:0.3 mL·min-1;柱温:25 ℃;进样量:2.0 µL。流动相及梯度洗脱程序,如表1所示。

表1 流动相梯度洗脱程序表
1.3.4 质谱条件
电喷雾电离正负离子模式:ESI(±);扫描方式:多反应监测(MRM);离子化温度:350 ℃;干燥器流量:10 L·min-1;喷雾电压(IS):+4000 V/-3500V; 雾化器压力:30 psi;监测离子及对应检测参数如表2所示。
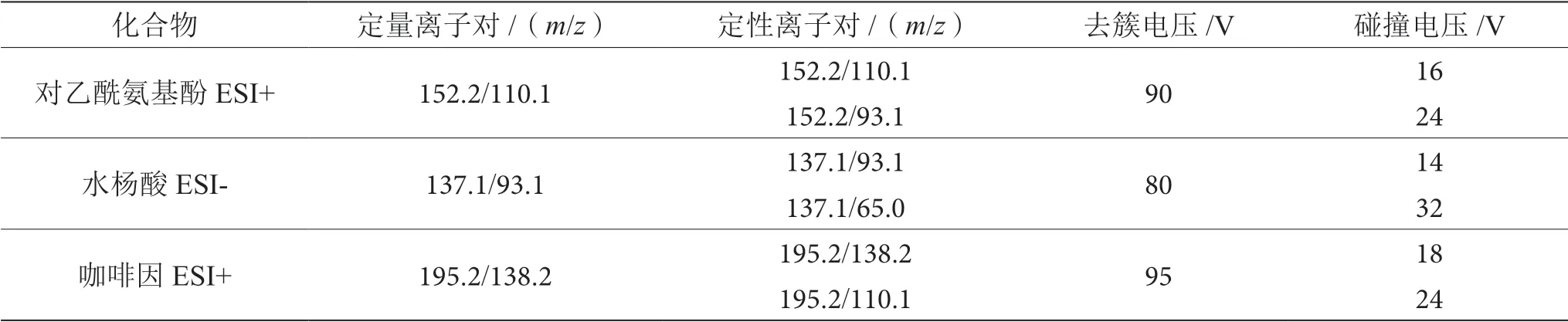
表2 监测离子及对应参数表
(1)定性测定。在相同的实验条件下测定试样溶液,若试样溶液质量色谱图中各物质的保留时间与标准溶液一致(变化范围在±2.5%以内),且试样定性离子的相对丰度与浓度相当的标准溶液中定性离子的相对丰度,其偏差不超过表3中规定,则可判定样品中存在该物质。

表3 定性离子相对丰度的最大允许偏差表
(2)定量测定。将试样溶液注入液相色谱-质谱/质谱仪中,得到各物质的定量离子峰面积,根据标准曲线计算试样溶液中各物质的浓度。
2 结果与分析
2.1 检出限与定量限的确定
将各组分标准物质逐级稀释,找到信噪比S/N=3时各组分浓度,结果确定对乙酰氨基酚、水杨酸、咖啡因的检出限分别为6 ng·mL-1、6 ng·mL-1、6 ng·mL-1,定 量 限(S/N=10)分 别 为20 ng·mL-1、20 ng·mL-1、 20 ng·mL-1。
2.2 标准曲线及相关系数
将配制好的标准系列溶液按照浓度由低到高的顺序进样测定,以各组分定量离子的色谱峰面积为纵坐标,以对应的浓度作为横坐标,得到标准曲线回归方程见图1。
由图1可得,对乙酰氨基酚标准曲线回归方程为y=180.486176x+529.528216,相关系数为0.99936;咖啡因标准曲线回归方程为y=53.704895x+50.217223,相关系数为0.99993;水杨酸标准曲线回归方程为y=11.006995x+21.250089,相关系数为0.99989。
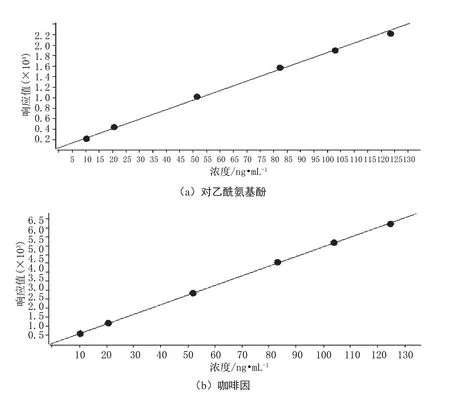
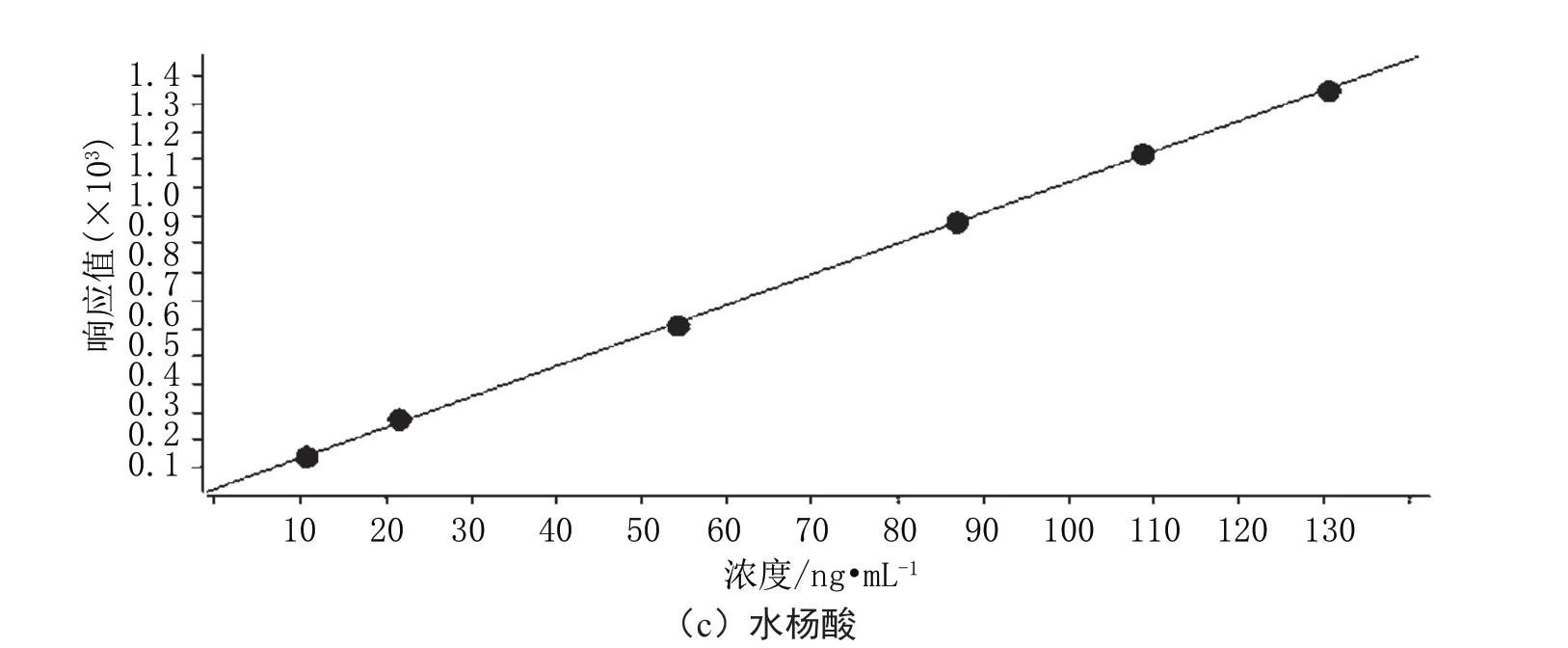
图1 标准曲线回归方程图
2.3 检测精密度及回收率
选用的蒸馏酒酒样基质均为空白基质,本底值均为未检出。
2.3.1 精密度
由表5可知,3种非法添加物质在空白基质中按照1倍、2倍、5倍定量限进行添加,样品经处理后上机检测,实验结果精密度均≤3.0%,表明该方法处理的样品含量结果精密度高。
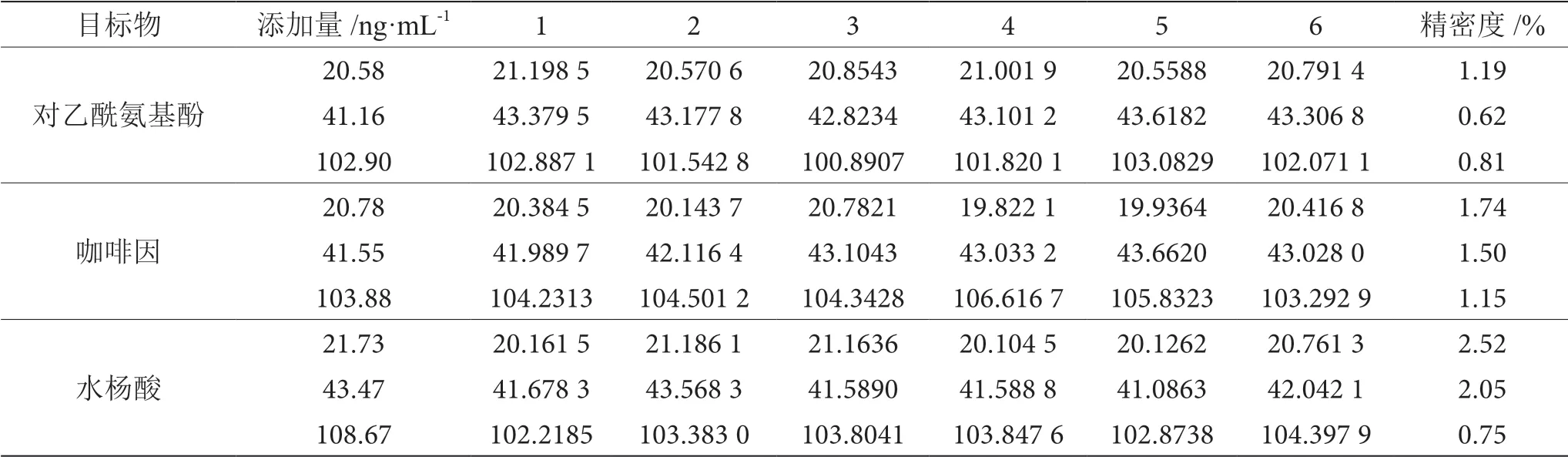
表5 对乙酰氨基酚、咖啡因、水杨酸检测结果表
2.3.2 回收率
由表6可知,3种物质在不同的加标浓度水平下的平均回收率为95.2%~105.0%。
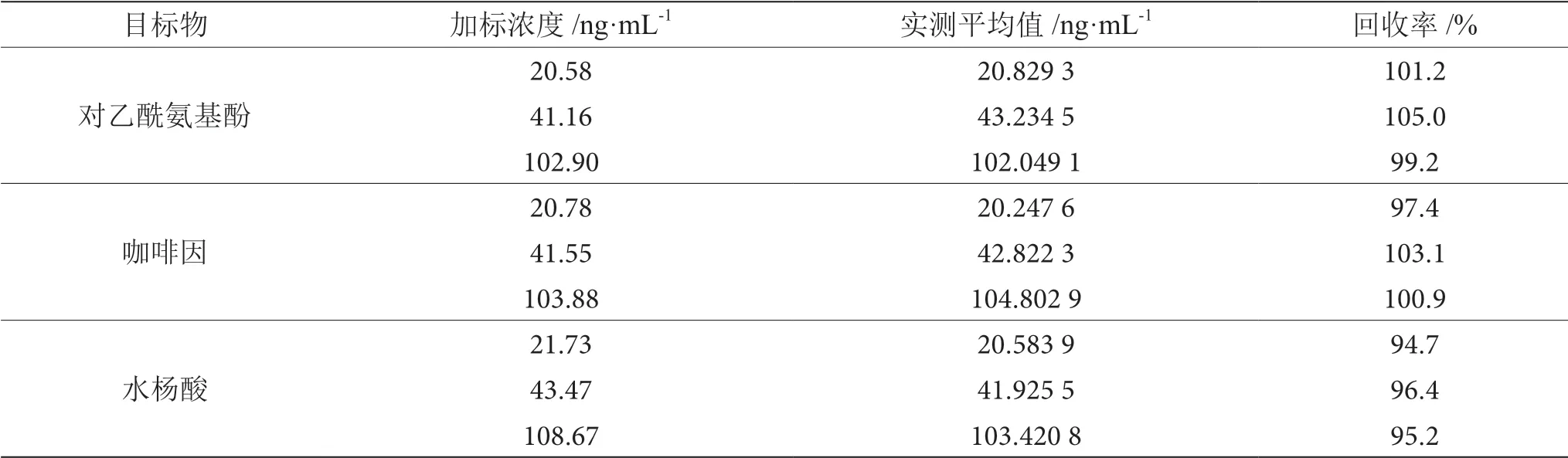
表6 对乙酰氨基酚、咖啡因、水杨酸回收率表
2.4 谱图
50 ng·mL-1对乙酰氨基酚、水杨酸、咖啡因标准溶液的总离子流色谱图见图2。
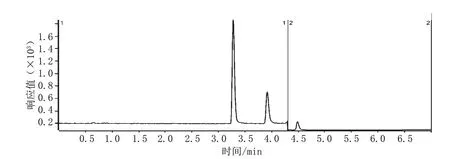
图2 50 ng·mL-1对乙酰氨基酚、水杨酸、咖啡因标准溶液的总离子流色谱图
50 ng·mL-1对乙酰氨基酚、水杨酸、咖啡因标准溶液的提取离子色谱图见图3。
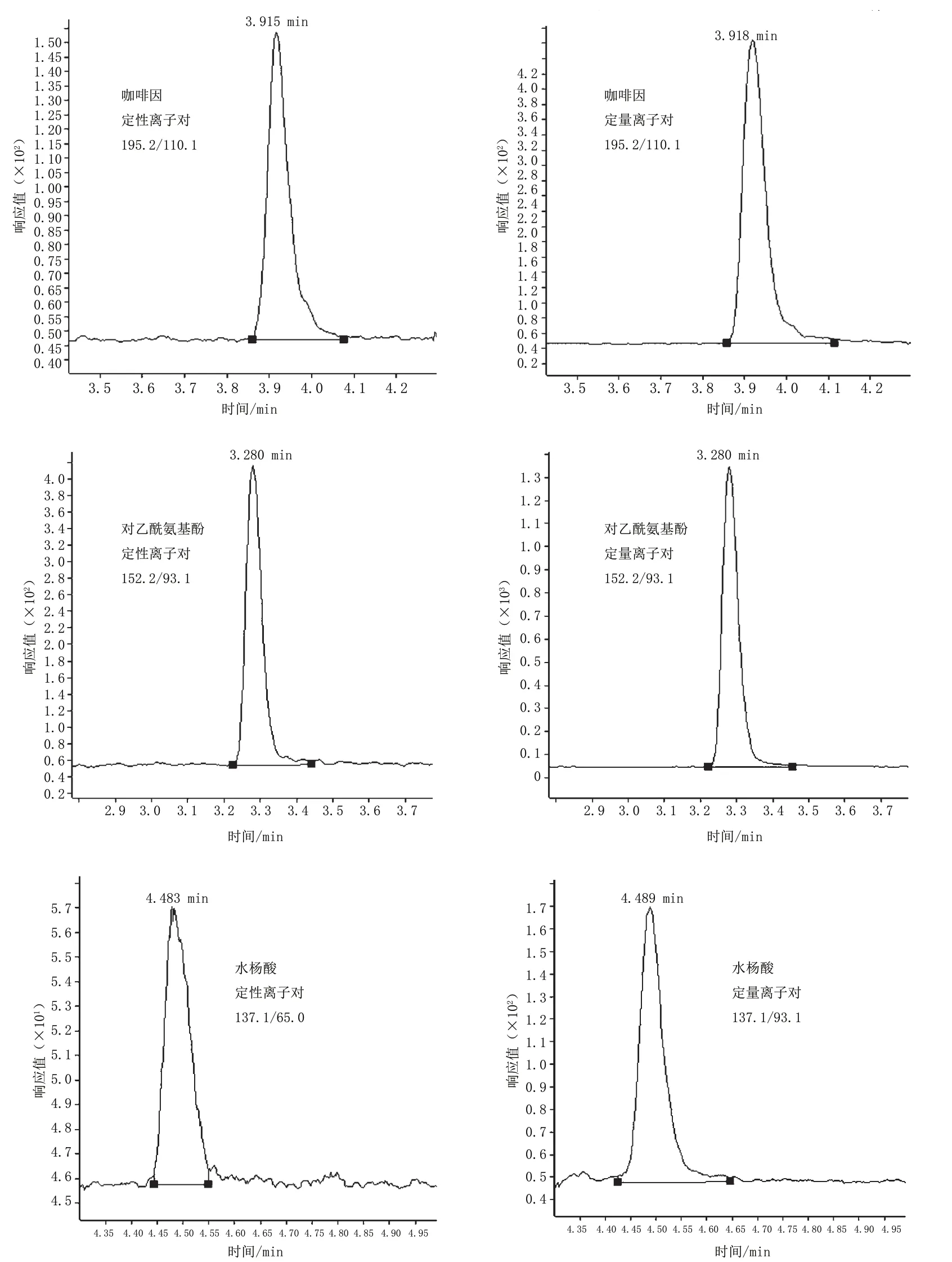
图3 50 ng·mL-1对乙酰氨基酚、水杨酸、咖啡因标准溶液的提取离子色谱图
2.5 样品测定结果
采用本检验方法对市售的20种白酒进行了检测,均未检出非法添加对乙酰氨基酚、水杨酸、咖啡因。
3 结论
蒸馏酒中非法添加物质的查处和检测能够完善市场监管需求。本文用超高效液相色谱-三重四极杆串联质谱法检测蒸馏酒中头痛粉的3种主要成分(包含水解成分),即对乙酰氨基酚、水杨酸、咖啡因。结果显示,该方法灵敏度、加标回收率高,重复性好,不仅对蒸馏酒中非法添加对乙酰氨基酚、水杨酸、咖啡因的分析检测方法起到了优化作用,还为食品安全的市场监管工作提供了准确有效的科学依据。
猜你喜欢
山西化工(2021年5期)2021-01-25 15:00:58
保健与生活(2020年4期)2020-03-02 02:27:36
天然产物研究与开发(2018年5期)2018-06-13 03:23:32
大自然探索(2017年10期)2017-10-28 06:47:59
大自然探索(2017年5期)2017-05-26 17:48:07
实用临床医学(2016年8期)2016-06-07 01:28:16
中国药物应用与监测(2015年5期)2015-12-11 03:15:53
化工进展(2015年6期)2015-11-13 00:27:14
结核与肺部疾病杂志(2015年3期)2015-07-18 11:08:56
应用化工(2014年11期)2014-08-16 15:59:13
